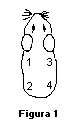
NORMA OFICIAL MEXICANA NOM-039-SSA1-1993, BIENES Y SERVICIOS. PRODUCTOS DE PERFUMERÍA Y BELLEZA. DETERMINACIÓN DE LOS ÍNDICES DE IRRITACIÓN OCULAR, PRIMARIA DÉRMICA Y SENSIBILIZACIÓN.
Al margen un sello con el Escudo Nacional, que dice: Estados Unidos Mexicanos.- Secretaría de Salud.
JOSE MELJEM MOCTEZUMA, Director General de Control Sanitario de Bienes y Servicios, por acuerdo del Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, con fundamento en los artículos 39 de la Ley Orgánica de la Administración Pública Federal; 38 fracción II, 47 de la Ley Federal sobre Metrología y Normalización; 8o. fracción IV y 13 fracción I del Reglamento Interior de la Secretaría de Salud.
PREFACIO
En la elaboración de esta Norma participaron los siguientes organismos e instituciones:
SECRETARIA DE SALUD
Laboratorio Nacional de Salud Pública
Dirección General de Control Sanitario de Bienes y Servicios
UNIVERSIDAD NACIONAL AUTONOMA DE MEXICO
Facultad de Química
AVON COSMETICS, S.A. DE C.V.
BEIERSDORF DE MEXICO, S.A. DE .C.V.
CAMARA NACIONAL DE LA INDUSTRIA DE LA PERFUMERIA Y
COSMETICA (CANIPEC)
CENTRO DERMATOLOGICO LADISLAO DE LA PASCUA
CLAIROL DE MEXICO, S.A.
LABORATORIOS ACONCA, S.A.
PROCTER & GAMBLE, S.A. DE C.V.
PRODUCTORA DE COSMETICOS, S.A. DE C.V.
REVLON, S.A.
YVES ROCHER DE MEXICO, S.A. DE C.V.
INDICE
0. INTRODUCCION
1. OBJETIVO Y CAMPO DE APLICACION
2. FUNDAMENTO
3. DEFINICIONES
4. SIMBOLOS Y ABREVIATURAS
5. PRUEBAS IN VIVO
6. PRUEBAS IN VITRO
7. MUESTREO
8. CONCORDANCIA CON NORMAS INTERNACIONALES
9. BIBLIOGRAFIA
10. OBSERVANCIA DE LA NORMA
11. VIGENCIA
12. APENDICE NORMATIVO
Apéndice A
13. APENDICE INFORMATIVO
Apéndice A
0. Introducción
Los productos de perfumería y belleza son formulaciones que contienen sustancias químicas e ingredientes que pueden causar lesiones en ojos o en la piel, por ser éstos de uso cotidiano es importante comprobar que su contenido sea inocuo. Con objeto de determinar si un producto es irritante o sensibilizante, las primeras pruebas se efectuaron en animales, como es la denominada Prueba de Draize. Estas pruebas in vivo demostraron que habían cambios visibles cuantificables. En la actualidad, además de éstas, se realizan pruebas de parche por inducción en humanos, ambas son útiles para evaluar los daños que puedan presentar dichos productos.
La continuidad en los estudios de los efectos tóxicos de los productos en general, han sido publicados en la última década, un número de aproximaciones de pruebas in vitro, cuyo objetivo es minimizar la necesidad de usar animales de prueba. Las pruebas in vitro se llevan a efecto por diferentes técnicas: unas son presentadas como pruebas de citotoxicidad, otras estudian la destrucción celular, inhibición del crecimiento, integridad o permeabilidad de la membrana y al final se media por el consumo celular de colorantes no tóxicos. La intensidad del teñido de la célula está directamente relacionado con el número de células sobrevivientes. Varios de los llamados sistemas intermedios se proponen como alternativos, lo que parece estar relacionado más íntimamente con el objetivo in vivo o aun usan órganos aislados, como el ojo de bovino. También se efectuan pruebas en huevos de gallina, donde la membrana corioalantoica se usa como modelo de membrana vascularizada.
Esta Norma establece los métodos para determinar las características de irritación y de sensibilización, con el fin de que los fabricantes puedan asegurar que sus productos sean "no irritantes" para el usuario y prevenir los daños a la salud que puedan ocasionar, al aplicarse o usarse directamente.
1. Objetivo y campo de aplicación
1.1 Esta Norma Oficial Mexicana establece los métodos de prueba para determinar los índices de irritación ocular, primaria dérmica y sensibilización, así como los límites máximos de aceptación.
1.2 Esta Norma Oficial Mexicana es de observancia obligatoria en el territorio nacional para las personas físicas o morales que requieran la determinación de estos índices.
2. Fundamento
Los animales se han utilizado para determinar la irritación ocular, primaria dérmica y la sensibilización en los productos de perfumería y belleza. Estas pruebas in vivo se efectúan poniendo la muestra del producto en contacto con el ojo o la piel del animal, para observar los cambios visibles y obtener los resultados haciendo las mediciones correspondientes. En la prueba del "parche" en humanos, de manera similar, se pone en contacto externo la muestra con la piel de la persona para medir la irritación dérmica y la sensibilización.
Las nuevas estrategias para determinar la seguridad de los cosméticos permiten escoger ensayos sensibles in vitro, pruebas confiables, reproducibles, prácticas y rutinarias, donde los parámetros de prueba se enfocan a los factores causales del daño en el tejido. Existen ahora un buen número de pruebas in vitro, que son alternativas potenciales para sustituir las pruebas en animales. Se han observado que los procesos agudos de irritación primaria en la piel y membranas mucosas se correlacionan bastante bien con los datos de diferentes sistemas de pruebas in vitro.
3. Definiciones
Para fines de esta Norma se entiende por:
3.1 Cepo, el aparato que sirve para sujetar e inmovilizar animales para experimentación.
3.2 Conjuntiva bulbar, la mucosa que tapiza la superficie anterior del ojo.
3.3 Conjuntiva palpebral, la mucosa que tapiza la cara posterior del párpado.
3.4 Córnea, sección anterior transparente del globo ocular.
3.5 Desafío, aplicación del mismo agente empleado en el proceso de inducción para verificar si es sensibilizante.
3.6 Edema, inflamación producida por acumulación excesiva de líquido seroalbuminoso en el tejido celular, debida a diversas causas.
3.7 Eritema, enrojecimiento difuso o en manchas de la piel, producido por la congestión de los capilares, debido a diversas causas.
3.8 Hemólisis, desintegración de los corpúsculos sanguíneos con liberación de hemoglobina.
3.9 Inducción, acción y efecto de inducir o causar una reacción por aplicación de un agente.
3.10 Irritación dérmica, alteración fisiológica de la piel provocada por algún agente físico, químico o biológico.
3.11 Irritación ocular, alteración fisiológica de las membranas oculares, provocada por algún agente físico, químico o biológico.
3.12 Membrana corioalantoica, capa celular extra embrionaria formada por la porción externa de la membrana alantoides, sobre las membranas amniótica y vitelina, que fija el embrión a la pared uterina, nutre y envuelve al huevo.
3.13 Membrana nictante, tejido delgado y flexible que envuelve y cubre el ojo.
3.14 Opacidad corneal, la turbidez de la transparencia corneal que se manifiesta en forma de una capa blanquecina.
3.15 Quemosis, engrosamiento de las conjuntivas ocular y palpebral y del tejido celular subyacente, de coloración rojiza más o menos acentuada, según el grado de inflamación.
3.16 Secreción, producto del funcionamiento fisiológico de una glándula; cuando la secreción elaborada es lanzada por un conducto excretor a otro órgano al exterior, es externa y cuando el producto segregado es vertido a la circulación sanguínea, es interna.
3.17 Sensibilización, proceso por el que las células se hacen más sensibles a la acción de un agente.
4. Símbolos y abreviaturas
Cuando en esta Norma se haga referencia a los siguientes símbolos y abreviaturas se entiende por:
ºC grados Celsius
g gramo
kg kilogramo
µl microlitro
mg miligramo
mg/ml miligramo por mililitro
ml mililitro
mm milímetro
mmol/l milimol por litro
M molar
nm nanómetro
p/v peso por volumen
% por ciento
rpm revoluciones por minuto
v/v volumen por volumen
Cuando en la presente Norma se mencione al Reglamento, debe entenderse que se trata del Reglamento de la Ley General de Salud en Materia de Control Sanitario de Actividades, Establecimientos, Productos y Servicios.
5. Disposiciones sanitarias
5.1 Los productos en los cuales se aplicarán los métodos objeto de esta Norma, se encuentran establecidos en el Reglamento. Adicionalmente, los fabricantes efectuarán estas pruebas en productos cuyos textos publicitarios incluyan las siguientes leyendas: "no irrita", "producto hipoalergénico".
5.2 En los casos de que los ingredientes de la fórmula sean los mismos con relación a otros productos del mismo fabricante, ya existentes y que cuenten con información sobre pruebas toxicológicas que documenten que son aceptables para su uso en humanos, el fabricante podrá presentar estas pruebas como justificación de que el nuevo producto es toxicológicamente adecuado para su uso.
5.3 Los fabricantes podrán efectuar métodos diferentes a los incluidos en esta Norma, para el control interno de sus productos; pero para efectos de comprobación de resultados por acciones de verificación sanitaria, se deben ajustar a los métodos de la presente Norma.
5.4 Las pruebas en humanos deben ser realizadas a través de protocolos éticamente desarrollados y supervisados por la Secretaría de Salud.
5.5 En el caso de estudios efectuados en el extranjero, serán aceptados si se llevan a cabo de acuerdo a las buenas prácticas clínicas establecidas en el país de origen. Esto debe ser documentado a través de un escrito certificado por una institución educativa, asociación científica reconocida o Autoridad Sanitaria del país de procedencia.
6. Pruebas in vivo
6.1 Prueba de Irritación Ocular
6.1.1 Materiales y reactivos
Una jeringa de 1 ml (sin aguja) o pipetas de 1 ml con divisiones de 0,10 ml o pipetas de 0,1 ml con divisiones de 0,01ml
Tres conejos albinos con un peso entre 2 y 3 kg con ojos sanos y sin defectos
Tres cepos
Agua destilada esterilizada
Un matraz Erlenmeyer de 125 ml esterilizado
Una lente de aumento
Solución salina al 0,9% esterilizada
Mortero
Tamiz del No. 40 (0,42 mm de abertura de malla)
Balanza analítica
6.1.2 Procedimiento
Esta prueba debe realizarse en 3 conejos. Sujetar los conejos en los cepos. Asimismo sujetar suavemente el párpado del conejo para mantener el ojo de prueba abierto. Aplicar directamente en la córnea 0,1 ml del líquido de prueba o 0,01 ml si se opta por la prueba con volumen bajo, utilizando la jeringa o la pipeta (utilizar una jeringa limpia para cada sustancia de prueba).
El otro ojo queda como testigo aplicando la misma cantidad de agua destilada y de la misma manera. En el caso de jabones y champúes deberá prepararse una solución al 10% (p/v o v/v), utilizando como vehículo agua destilada: aplicar 0,1 ml o 0,01 ml de esta solución de la manera antes descrita.
Para sólidos, el material de prueba deberá ser pulverizado con ayuda de un mortero, tamizado y con éste hacer una suspensión acuosa al 10%. Aplicar 0,1 ml o 0,01 ml de esta suspensión directamente en la córnea del ojo de prueba, quedando el otro ojo como testigo.
En el caso de pastas, calcular la densidad en mg/ml, dividir 0,1 g o 0,01 g entre la densidad y la cantidad resultante del material de prueba, se aplica directamente en la córnea del ojo.
Liberar el párpado inmediatamente después de la aplicación, sin forzar el parpadeo ni manipular.
Llevar un registro de la hora de aplicación. Y anotar además si los animales de prueba mostraron cualquier signo de malestar como consecuencia de la aplicación de la sustancia de prueba.
Mantener a los animales en sus cepos durante tres horas y después regresarlos a sus jaulas.
6.1.3 Evaluación de la prueba
6.1.3.1 Observar los ojos de los conejos a la 1a., 2a. y 3a. hora, a las 24 y 48 horas y al 4o., 5o., 6o. y 7o. días después de aplicada la sustancia de prueba, usando el ojo no tratado como control o testigo, haciéndose las anotaciones correspondientes.
6.1.3.2 La prueba podrá suspenderse después del 3er. día, siempre y cuando no se observe irritación alguna en los ojos de prueba.
6.1.3.3 Evaluar en los ojos de prueba la reacción que ante la aplicación de la muestra presentan la conjuntiva, la córnea y el iris, con ayuda de una lente de aumento, utilizando la siguiente escala de calificación.
6.1.4 Escala para la evaluación numérica de las lesiones oculares
Observar en el ojo cerrado la quemosis y lacrimación, abrir el párpado para evaluar la conjuntiva, la córnea y el iris, utilizando la lente de aumento.
6.1.4.1 Conjuntiva
Enrojecimiento (se refiere a la conjuntiva palpebral y bulbar, excluyendo la córnea y el iris)
Vasos normales 0
Vasos capilares con ligero enrojecimiento 1
Enrojecimiento 2
Enrojecimiento difuso intenso 3
Quemosis
No hay inflamación 0
Inflamación ligera, incluyendo la membrana nictante 1
Inflamación con eversión parcial del párpado 2
Inflamación con párpados cerrados a la mitad 3
Inflamación con párpados totalmente cerrados 4
Secreción
Ausencia de secreción 0
Secreción ligera apenas perceptible 1
Secreción con humedecimiento de los párpados y del pelo adyacente al borde palpebral externo 2
Secreción con humedecimiento de los párpados y del pelo sobre grandes zonas alrededor del ojo 3
Calificación total
(a + b + c) x 2 = 20 (máximo)
Donde:
a = Valor del enrojecimiento
b = Valor de la quemosis
c = Valor de la secreción
6.1.4.2 Córnea
Grado de opacidad
No hay opacidad (no hay pérdida de brillantez o luminosidad) 0
Ligera opacidad (sin perder la transparencia pero sí la brillantez) 1
Presencia de opacidad aún traslúcida con el iris ligeramente obscurecido 2
Presencia de opacidad con iris poco visible y contorno de la pupila dificilmente visible 3
Presencia de opacidad que hace al iris invisible 4
Areas de opacidad
Menos de un cuarto 1
Entre un cuarto hasta la mitad 2
Más de la mitad y tres cuartos 3
De tres cuartos a toda el área 4
Total de lesiones en córnea
a x b x 5 = 80 (máximo)
Donde:
a = Valor del grado de opacidad
b = Valor del área de opacidad
6.1.4.3 Iris
Valores
Normal 0
Congestionado, con inyecciones circuncorneales y obviamente más arrugado que lo normal (una o varias de estas características), con el iris aún reaccionando a la luz (una reacción lenta es una reacción positiva) 1
No hay reacción a la luz, hemorragia, lesión considerable (una o varias de estas características) 2
Total de lesiones en iris
a x 5 = 10 (máxima)
Donde:
a = Valor de la lesión en iris
6.1.5 Interpretación de los resultados
Se suman las calificaciones obtenidas en conjuntiva, córnea e iris para cada conejo, se promedian los valores de los 3 conejos, obteniéndose así la calificación de la prueba por día.
Una vez terminada la prueba, según lo mencionado en el párrafo 6.1.3.2 e incluyendo el valor del último día de la prueba, se promedian los valores de las calificaciones de la prueba por día.
El valor promedio obtenido se divide entre 110 (suma total de las calificaciones máximas posibles) y dependiendo del cociente obtenido se clasificará el producto de acuerdo a la siguiente tabla:
0,0 a 0,1 no irritante
Más de 0,1 a 0,3 ligeramente irritante
Más de 0,3 a 0,5 irritante
Más de 0,5 a 1,0 irritante severo
6.1.6 Criterio de aceptación
El producto que tenga una calificación no mayor de 0,3 será aceptado como producto apto para uso humano. En el caso de productos destinados a bebés la calificación máxima deberá ser 0,1.
6.2 Prueba de irritación en piel de conejos
6.2.1 Materiales y reactivos
3 conejos albinos de 2,0 a 3,5 kg de peso, cualquier sexo y sin daño en la piel
3 cepos
3 jaulas individuales de malla metálica
Máquina rasuradora eléctrica, para animales pequeños, con peine del No.40 y del No. 0
Pipeta de 1 ml graduada en centésimas
Matraces Erlenmeyer
Matraces aforados de 50 ml
Agua destilada esterilizada
Fuente de luz blanca mínimo de 60 watts
Lente de aumento
Gasa quirúrgica esterilizada
Tela adhesiva
Tela permeable
Mortero con pistilo
Balanza analítica
Tamiz del No. 40 (0,42 mm de abertura de malla)
La cantidad de sustancia de prueba para cada tipo de productos se especifica en la tabla 1.
6.2.2 Procedimiento
6.2.2.1 Preparación de los animales: el día anterior a la realización de la prueba, se rasura el dorso de los conejos de manera que quede sin pelo desde la región escapular a la lumbar a un lado y otro de la columna vertebral. Para ello utilizar primero el peine del No. 40 y después el del No. 0. Esto debe hacerse con mucho cuidado, a fin de no lesionar la piel al rasurar.
6.2.2.2 El material de prueba se aplicará directamente en el dorso de cada animal, cubrir con un parche de gasa quirúrgica de 2 x 2 cm, con un grosor de 4 a 8 monocapas, colocado en el sitio elegido. Considerar como control cualquier otra área de la piel en esta zona.
6.2.2.3 Colocar a cada animal en un cepo para minimizar sus movimientos durante la prueba. Si la sustancia de prueba es un líquido, aplicar la cantidad que se indica en la tabla 1 directaménte en la piel, después, sujetar la gasa al conejo con una tela adhesiva. En el caso de materiales sólidos, semisólidos o en polvo, sujetar el parche a la piel en uno de los lados utilizando tela adhesiva; a continuación colocar el material de prueba en la cantidad que se indica en la tabla 1, entre el parche y la piel, posteriormente cerrar el lado restante con tela adhesiva.
Para sólidos y polvos, el producto deberá molerse y tamizarse antes de pesarse. Cuando el material de prueba contenga sustancias volátiles, se deberá permitir la evaporación del disolvente antes de aplicar el parche con la sustancia de prueba y de colocarlo en la piel del conejo.
El tiempo de aplicación del parche varía dependiendo de la naturaleza del producto; este tiempo para cada grupo de productos se especifica en la tabla 1.
6.2.3 Evaluación de la prueba
Una vez que ha transcurrido el tiempo de aplicación, sacar a los conejos de los cepos, remover el parche y eliminar los restos del material de prueba con una toalla húmeda. Regresar cada animal a su jaula.
Después de haber removido los parches, evaluar el sitio de prueba de acuerdo con las siguientes escalas:
Eritema
0 No eritema
1 Eritema ligero, apenas perceptible
2 Eritema bien definido
3 Eritema de moderado a severo
4 Eritema severo (rojo betabel)
Calificación máxima posible de eritema: 4
Edema
0 No edema
1 Edema ligero apenas perceptible
2 Edema ligero con bordes sobresalientes con elevación definida
3 Edema moderado, con una elevación máxima de 1 mm, aproximadamente
4 Edema severo, elevación mayor de 1 mm extendiéndose más allá del sitio de aplicación
Calificación máxima posible de edema: 4
Las evaluaciones deberán realizarse a los 30 o 60 minutos después de remover el parche, a las 24 horas y a las 72 horas de iniciada la prueba.
6.2.4 Análisis de datos
Una vez obtenidas las lecturas de Eritema y Edema, promediar las calificaciones de eritema de las 3 evaluaciones (1/2 o 1 hora, 24 y 72 horas después de remover el parche) para los tres animales (9 calificaciones en total) y por separado promediar las calificaciones de edema de la misma manera (9 calificaciones en total).
A partir de estos promedios, calcular el índice de irritación primaria como sigue:
Indice de irritación primaria = Promedio de eritema + Promedio de edema.
6.2.5 Interpretación de los resultados
Con base en el Indice de Irritación Primaria, los productos podrán ubicarse en alguna de las siguientes categorías:
0 - 1 No irritante
1,1 - 2 Ligeramente irritante
2,1 - 5 Moderadamente irritante
5,1 - 6 Irritante moderado a severo
6,1 - 8 Irritante severo
6.2.6 Criterio de aceptación
Todos los productos para uso en bebés deberán tener un índice de irritación primaria no mayor de 2.
El producto que tenga un índice de irritación primaria no mayor de 5 será aceptado como un producto apto para uso en adultos.
Cuando un producto obtenga una calificación entre 5,1 y 6 deberá optar por cualquiera de las tres alternativas siguientes:
6.2.6.1 Ostentar la leyenda "Precaución, este producto es irritante".
6.2.6.2 Repetir la prueba en animales vírgenes, si los resultados de esta segunda prueba confirman los obtenidos en la primera, deberá optarse por la alternativa 6.2.6.1.
Si el índice de irritación primaria obtenido en la segunda prueba tiene un valor aceptable conforme a lo que se establece en el punto 6.2.6, el producto será considerado apto para uso humano.
Los productos que tengan una calificación mayor de 6 no serán aceptados para su uso en humanos.
6.2.6.3 Proceder a realizar la prueba en humanos, descrita en el punto 6.3.
TABLA 1
PREPARACION DE LA MUESTRA
PRODUCTO PREPARACION DE LA CANTIDAD A CONDICIONES DE
MUESTRA APLICAR APLICACION
Champú, jabón líquido, 10% sol. acuosa 0,3 ml Parche semiocluido 4
acondicionador horas.
enjuague, jabón, crema y
espuma de afeitar, jabón
de tocador, jabón
desodorante, productos
para baño o ducha. horas.
Loción capilar Presentación original 0,3 ml Dejar evaporar por 15
min. Parche semiocluido
4 horas.
Perfume, loción y Presentación original 0,3 ml Dejar evaporar por 15
similares min. Parche semiocluido
24 horas.
Endurecedor, adhesivo y Presentación original Lo suficiente para Dejar evaporar por 15
reparador de uñas cubrir 1 cm2 del min. Parche semiocluido
parche. 4 horas.
Removedor de cutícula Presentación original 0,3 ml o g Parche semiocluido 1
hora.
Sombra, crema, Presentación original 0,3 g Parche semiocluido 24
maquillaje, talco, polvo, horas.
lápiz labial
Desmaquillante, Presentación original 0,3 g Dejar evaporar por 15
delineador, máscara min. Parche semiocluido
para pestañas, por 24 horas.
bronceador, filtro solar
Tinte Presentación original 0,3 ml Parche semiocluido 1
hora.
Ondulante, fijador Presentación original 0,3 ml Parche semiocluido 1
permanente hora.
Desodorante y Presentación original 0,3 g o ml Parche semiocluido 24
antitranspirante horas.
Depilatorio Presentación original 0,3 g Parche semiocluido 1
hora.
6.3 Prueba de irritación en piel de humanos (Prueba del parche con tres aplicaciones)
6.3.1 Materiales y reactivos
Un mínimo de 10 voluntarios, los cuales, antes de comenzar su participación en el estudio, deben proporcionar al investigador una carta donde informen por escrito su consentimiento para participar voluntariamente en la prueba (de acuerdo al formato que se encuentra en el apéndice informativo A 1). El investigador proporcionará a cada voluntario la información necesaria acerca del propósito y naturaleza del estudio, cada sujeto recibirá una copia del informe de consentimiento.
Matraces aforados
Pipetas volumétricas
Agua destilada esterilizada
Fuente de luz, lámpara incandescente de 100 watts de luz de día con bulbo azul
Lente de aumento
Matraces Erlenmeyer
Parches de gasa quirúrgica esterilizada de un grosor de 4 monocapas, de 2 cm por lado
Micropore u otra cinta adhesiva porosa similar utilizada en la práctica médica
Mortero y pistilo
Balanza analítica
Tamiz No. 40 (0,42 mm de abertura de malla)
Alcohol etílico
6.3.2 Elección de los voluntarios
Para que un voluntario pueda participar en esta prueba, debe cumplir con los siguientes requisitos:
Que sea una persona sana.
Que haya transcurrido por lo menos un mes desde su última participación en una prueba de parche.
Que tenga por lo menos 18 años. No más del 20% de los participantes podrán tener más de 65 años.
Un voluntario NO podrá participar en esta prueba por cualquiera de las siguientes razones:
Si ha sido sometido a algún transplante de órgano que requiere el uso de medicamentos inmunodepresores (a excepción de transplante de córnea).
Si presenta psoriasis, eczema u otro tipo de erupciones en cualquier sitio de la piel, cáncer en la piel o alguna enfermedad de ésta, que pudiera interferir con las evaluaciones efectuadas en este estudio, o que pudieran exponer al voluntario a un riesgo inaceptable.
Si el voluntario está tomando algún medicamento esteroide anti-inflamatorio por administración sistémica, o si se está aplicando cualquier medicamento en el sitio de aplicación del parche.
Si presenta diabetes y está tomando insulina.
Si presenta asma severo o algún tipo de alergia respiratoria que requiere de una terapia crónica o frecuente con administración de medicamentos.
Si se le ha practicado mastectomía bilateral o unilateral durante el último año, o extirpación de nódulos linfáticos axilares.
Si está recibiendo tratamiento para cualquier tipo de cáncer, o si ha sido tratado contra cáncer durante los últimos 6 meses.
Si presenta alguna enfermedad de inmunodeficiencia (lupus, tiroiditis, etc.).
Si se desarrolló alergia como consecuencia de su participación en pruebas de parche previas.
Si la persona se encuentra embarazada o en periodo de lactancia.
Si la persona presenta antecedentes de trastornos mentales.
La elección de los voluntarios será evaluada a través de un cuestionario (que se encuentra en el Apéndice informativo A 2) que debe ser llenado por cada persona. Antes de comenzar su participación en el estudio, los voluntarios que sean elegidos en esta prueba deben proporcionar al investigador una carta en la que informen por escrito su consentimiento a participar voluntariamente en la prueba (Apéndice informativo A 1). El investigador debe proporcionar a cada voluntario la información necesaria acerca del propósito y naturaleza del estudio, cada sujeto recibirá una copia del informe de consentimiento.
6.3.3 Preparación de la sustancia de prueba
Los productos que el usuario emplea tal como se presentan a la venta, se aplican directamente sobre la piel. Los que se emplean en polvo (maquillajes, sombras de ojos, etc.) se aplican como polvos al realizar esta prueba. En el caso de los que el usuario emplea diluidos, la concentración del material de prueba debe ser aplicada en el parche de acuerdo a lo que se indica en la Tabla 1.
En el caso de los sólidos, éstos deben ser pulverizados previamente utilizando un mortero y después tamizados (tamiz del No. 40). En el caso de los líquidos, se preparará la concentración adecuada utilizando agua destilada (ver Tabla 1).
Se utilizan parches de algodón o similares a los utilizados en la práctica médica, aplicar la cantidad de sustancia de prueba especificada en la Tabla 1. Debe tenerse cuidado de no aplicar una cantidad excesiva de la sustancia, ya que podría escurrir o abarcar un área mayor que la del parche.
6.3.4 Procedimiento
Se pueden aplicar hasta 4 parches por brazo y sólo en caso de mucha necesidad un total de 8 parches por voluntario, con un espacio mínimo entre los bordes de cada parche de 2 cm.
Los parches se aplican en la superficie lateral del brazo entre el hombro y el codo, con el brazo en una posición relajada al lado del cuerpo. Los lugares de aplicación de los parches se eligen aleatoriamente y se deben rotar entre los diferentes voluntarios para minimizar la variación entre los sitios, como en el siguiente ejemplo:
4 muestras: 1-2-3-4, 2-3-4-1, 3-4-1-2, 4-1-2-3
3 muestras: 1-2-3, 2-3-1, 3-1-2
2 muestras: 1-2, 2-1
El sitio de aplicación para cada muestra y para cada sujeto voluntario, se anota claramente, conservándose su localización durante todo el tiempo que dure la prueba. Los sitios de prueba son marcados con 2 puntos arriba y 1 abajo con una solución al 0,5% de violeta de genciana, de forma que la localización del sitio de aplicación sea claramente visible para las subsecuentes evaluaciones y nuevas reaplicaciones del parche.
Los parches se aplican 3 veces, cada uno durante el tiempo que se especifica en la tabla 1 por 24 horas y después se remueven. Los sitios se evalúan 48 horas después de cada aplicación (72 horas después de un fin de semana).
Los parches se aplican y se evalúan de la siguiente manera:
Primer día: Se aplican los parches y se mantienen en su lugar el tiempo que se especifica en la Tabla 1, cubriéndose con material adhesivo como micropore, dicho material debe colocarse de manera que se ejerza igual presión sobre todo el parche.
Segundo día: Se instruye al voluntario para que él mismo se retire el parche y elimine cualquier residuo de la sustancia de prueba utilizando una toalla o un trozo de algodón húmedos.
Tercer día: Acudir al centro de prueba donde se realiza la evaluación del área expuesta, empleando una fuente luminosa artificial, de ser necesario, auxiliarse con una lente de aumento. Al terminar la evaluación se aplica el siguiente parche.
Cuarto día: Proceder como el segundo día.
Quinto día: Proceder como el tercer día.
Sexto día: Proceder como el segundo día.
Séptimo día: Proceder como el tercer día sin aplicar nuevo parche.
Las evaluaciones se califican utilizando la siguiente escala:
6.3.5 Escala de evaluación
0 No se presenta reacción visible en la piel
0,5 Mayor que 0 y menor que 1
1 Eritema ligero definido, sin erupciones ni grietas en la piel o bien ausencia de eritema con presencia de resequedad
1,5 Mayor que 1 y menor que 2
2 Eritema moderado, puede presentarse algunas pápulas o fisuras profundas y presentarse eritema moderado o severo en las grietas
2,5 Mayor que 2 y menor que 3
3 Eritema severo (color rojo brillante), pueden presentarse pápulas generalizadas o eritema severo con edema ligero (bordes bien definidos y elevados)
3,5 Mayor que 3 y menor que 4
4 Vesículas generalizadas o eritema moderado a severo o edema que se extiende más allá del área del parche
Los voluntarios deben ser evaluados por la misma persona durante toda la prueba.
Los parches no se aplican en el sitio que ha presentado una calificación igual o mayor de 2. Esta calificación se registra adecuadamente. La reaplicación del parche se hará en la parte anterior al sitio inicial de aplicación, si este sitio obtiene una calificación igual o mayor de 2, el tercer sitio se eligirá en la parte posterior al sitio inicial.
Si se presenta una irritación excesiva (calificación >2) en todos los participantes en la primera evaluación, para las aplicaciones subsecuentes debe disminuirse la concentración de la sustancia de prueba a la mitad para los productos que se aplicaron diluidos, o se disminuirá el tiempo de aplicación de esta sustancia a la mitad para aquellos productos que se aplican en su presentación original (ver Tabla 1). En cualquier caso el parche se reaplica en la parte anterior al sitio inicial de aplicación y, si se requiere, el tercer sitio se eligirá en la parte posterior al sitio inicial. Esto debe documentarse CLARAMENTE y tomarse en cuenta en los criterios de aceptación del producto.
6.3.6 Análisis de datos
En el caso de que obtenga una calificación de 3 o más, no se reaplica parche y se supone una calificación de 3 para las evaluaciones restantes.
Durante la prueba se pueden presentar como respuestas esperadas, reacciones que van de medio a moderado eritema, con alguna comezón o sensación de ardor. Los voluntarios deben de tener la forma de comunicarse (proporcionar un número telefónico) con el centro de prueba, tan pronto como la respuesta sea de mayor intensidad, a fin de que cuente con asistencia médica las 24 horas del día.
Cualquier sujeto que falte a una aplicación o evaluación debe ser eliminado del estudio. La razón por la cual se eliminó algún voluntario debe ser anotada para tomarse en cuenta al hacer la evaluación final.
6.3.7 Interpretación de los resultados
Después que han completado todas las sesiones de evaluación se suman las calificaciones obtenidas por día y se dividen entre el número total de sujetos, lo que representa al promedio de calificación para este día. El promedio de estas calificaciones es el INDICE DE IRRITACION PROMEDIO FINAL.
6.3.8 Criterios de aceptación
Los productos para uso en bebés deben presentar un índice de irritación promedio final no mayor de 1.
Los productos que tengan una calificación menor o igual a 1,5 serán aceptados como productos aptos para uso humano.
6.4 Prueba de sensibilización
Esta prueba podrá realizarse en piel de cobayos o en piel de humanos.
6.4.1 Prueba de sensibilización en piel de cobayos
6.4.1.1 Materiales y reactivos
En esta prueba deben utilizarse cobayos de la cepa Hartley, con un peso entre 300 y 400 g, para cada fase de la prueba se aconseja emplear el mismo número de hembras que de machos. El número de animales necesarios en cada fase es el siguiente:
Un mínimo de 10 animales de prueba (de preferencia 20) para las fases de inducción y desafío
10 animales control para la fase de redesafío, cuando ésta se requiera
4 animales para el estudio de irritación, previo a la fase de inducción
8 animales para el estudio de irritación, previo a la fase de desafío
Cepos dotados de una banda elástica de hule, para mantener sujeto al animal durante el tiempo de aplicación de los parches
Jaulas individuales de malla metálica u otro material adecuado
Rasuradora eléctrica, equipo para animales pequeños, con peines del No. 40 y del No. 0
Pipetas de volumen ajustable, graduadas en décimas de ml
Agua destilada esterilizada
Balanza analítica
Fuente de luz blanca fluorescente de 160 watts
Gasa quirúrgica esterilizada
Tela adhesiva (de adhesión suave)
Tamiz del No. 40 (0,42 mm de abertura de malla)
Bandas elásticas (tensoplast) quirúrgicas
6.4.1.2 Procedimiento
Consideraciones generales
El único periodo de estrés a que deben ser sometidos los animales es el tiempo que se les mantiene en los cepos, durante la exposición a la sustancia de prueba. La administración de analgésicos o sedantes puede modificar la respuesta inflamatoria a evaluar.
También puede modificarse el tono muscular, trayendo como consecuencia que las condiciones de oclusión de los parches se modifiquen de manera importante.
Por lo tanto, no se debe administrar este tipo de sustancias ni otros medicamentos a los animales durante el tiempo que dura la prueba.
6.4.1.3 Métodos de tratamiento y observación
6.4.1.3.1 El día anterior a la prueba debe rasurarse el dorso del animal en el área de aplicación de los parches con la rasuradora eléctrica, utilizando primero el peine del No. 40 y después el del No. 0. Esto debe hacerse cuidadosamente, sin lesionar la piel al rasurar.
6.4.1.3.2 Los parches a aplicar deben ser ocluidos, preparándose como sigue: El material de prueba se aplicará directamente al animal, cubrir con el parche de gasa quirúrgica esterilizada de 2 x 2 cm, con un grosor de 4 a 8 monocapas, colocado en cualquier sitio del dorso del animal. Considerar como control cualquier otra área de la piel en esta zona. En el caso de líquidos, aplicar 0,4 ml, en el caso de sólidos, polvos o pastas sujetar el parche a la piel en uno de los lados utilizando tela adhesiva, colocar el material de prueba 0,4 g sobre la piel, y cerrar el lado restante del parche con tela adhesiva. Los sólidos y polvos deberán ser molidos y tamizados antes de pesar.
Cuando el material de prueba contenga sustancias volátiles, se debe permitir la evaporación del disolvente antes de aplicar el material de prueba con la sustancia y de colocarlo en la piel del animal.
6.4.1.3.3 Colocar el animal en el cepo y aplicar el parche en el área adecuada. Ocluir el parche utilizando la banda elástica, ajustándola de manera que haga contacto con la región dorsal del animal de manera uniforme. Ajustar el cepo para minimizar los movimientos del animal durante el periodo de exposición.
Mientras permanezcan en el cepo, los animales deberán ser revisados cada 60 o 90 minutos, anotándose cuidadosamente cualquier síntoma de molestia. Los parches deberán permanecer por 6 horas en todas las fases de la prueba. Transcurrido este tiempo, remover el parche y limpiar el área de aplicación con una toalla o gasa húmeda y regresar los animales a su jaula.
6.4.1.4 Evaluación de la prueba
Se debe realizar la evaluación a las 18 horas, utilizando la escala que se incluye en la Tabla 2.
Repetir la evaluación 24 horas más tarde (es decir 48 horas después de haber removido el parche).
Debe utilizarse esta escala para realizar las evaluaciones de las pruebas preliminares de irritación y de la fase de desafío. La evaluación de todos los animales debe realizarse colocándolos bajo una fuente de luz blanca fluorescente de 160 watts, a una distancia de 90 cm y sobre un fondo negro plano. Esta evaluación debe hacerse por dos técnicos, los cuales deben estar de acuerdo en la calificación que se registre.
Además, cuando la respuesta sea difícil de evaluar pueden compararse los sitios de aplicación del parche de otros animales en el grupo de prueba y en el control antes de emitir la calificación.
6.4.1.5 Estudio de irritación previo a la fase de inducción
Efectuar un estudio previo a la fase de inducción, con objeto de determinar el potencial de irritación de la sustancia de prueba. Para ello utilizar un grupo de 4 cobayos, de preferencia 2 hembras y 2 machos.
Rasurar el pelo en el lomo y los flancos de cada animal, un día antes de iniciar la prueba.
Transcurridas 24 horas, aplicar los parches como se indicó en la 6.4.1.3.2, utilizando 4 concentraciones de la sustancia, incluido el 100%. Para hacer diluciones deberá utilizarse un vehículo adecuado (agua, aceite mineral, alcohol, etc.), dependiendo de la naturaleza de la sustancia de prueba. Los sitios de aplicación del parche en cada animal para esta fase del estudio deberán ser los indicados en la figura 1.
Deben alternarse los sitios de aplicación de las diferentes concentraciones entre los 4 animales, como se indica en la figura 2, para minimizar la variación de la respuesta entre sitios.
El sitio de aplicación de cada concentración en el animal deberá anotarse claramente. Los parches deberán permanecer aplicados durante 6 horas, después se remueven de acuerdo a lo indicado en el punto 6.4.1.3.3.
Evaluar la reacción de cada sitio de prueba a las 24 y 48 horas como se explica en el punto 6.4.1.4, utilizando la escala de evaluación (Tabla 2).
Elegir la concentración que provoque una irritación dérmica ligera, para realizar la fase de inducción.
Si como resultado de esa prueba se encuentra que la máxima concentración no produce irritación, esta concentración será elegida para realizar la fase de inducción y la fase de desafío. En aquellos casos en que una de las concentraciones utilizadas provoca una irritación dérmica ligera, será necesario realizar otro estudio de irritación previo a la fase de desafío, como se describe a continuación.
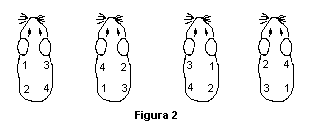
6.4.1.6 Estudio de irritación previo a la fase de desafío
Cuando sea necesario, realizar un nuevo estudio de irritación previo a la fase de desafío, considerando la concentración que provocó una irritación dérmica ligera como concentración máxima. A partir de este valor, elegir otras 3 concentraciones menores a intervalos regulares. En este caso sólo podrán aplicarse 2 parches en cada animal, como se muestra en la figura 3.
Alternar los sitios de aplicación entre los 8 animales, como se indica en la figura 4, para minimizar la variación entre sitios.
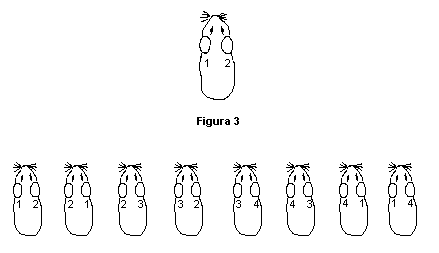
Aplicar los parches de la manera antes descrita, durante 6 horas. Transcurrido este tiempo, remover el parche y limpiar el área de aplicación de acuerdo a lo mencionado en el punto 6.4.1.3.3.
Evaluar a las 24 y 48 horas, utilizando la escala incluida en la Tabla 2.
Elegir la máxima concentración que NO provoca irritación para realizar la fase de desafío.
6.4.1.7 Fase de inducción
Emplear un mínimo de 10 cobayos como animales de prueba.
Día 0: Rasurar el pelo del flanco superior izquierdo de cada animal del grupo, como se describió en el punto 6.4.1.3.1.
Día 1: Aplicar los parches a los animales de acuerdo a lo descrito en el punto 6.4.1.3.2. El sitio de aplicación del parche de inducción en cada animal se indica en la figura 5.

Después de 6 horas de aplicado el parche, removerlo, limpiar como lo indica el punto 6.4.1.3.3 y regresarlo a su jaula.
Días 6 y 13: Rasurar nuevamente a los animales.
Días 7 y 14: Proceder como el día 1.
Anotar cualquier reacción de la piel en el sitio de aplicación del parche durante esta fase.
6.4.1.8 Fase de desafío
Día 27: Rasurar el flanco posterior izquierdo de los 10 (o 20) cobayos de la fase de inducción y a 10 animales no expuestos a sustancias de prueba, que servirán como control.
Día 28: Aplicar un parche de desafío a cada uno de los 20 (o 30) animales (10 o 20 de prueba y 10 de control) como se describe en el punto 6.4.1.3.2, en el sitio que se indica en la figura 6.
Note que este sitio es DIFERENTE al empleado para aplicar el parche de inducción.
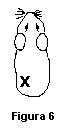
Remover el parche después de 6 horas de aplicación, limpiar el sitio y regresar cada animal a su jaula.
Día 29: Evaluar la respuesta a las 24 horas de haber removido el parche de desafío, utilizando la escala incluida en la Tabla 2.
Día 30: Evaluar la respuesta a las 48 horas de haber removido el parche de desafío. Proceder como el día 29.
6.4.1.9 Fase de re-desafío
Cuando la respuesta obtenida en la fase de desafío es dudosa, deberá realizarse un re-desafío en el grupo de prueba de 7 a 15 días después del primer desafío.
Deberán incluirse otros 10 cobayos no expuestos anteriomente a la sustancia de prueba como control.
Rasurarse la porción dorsal derecha, como se indica en el punto 6.4.1.3.1, en el sitio indicado en la figura 7.
Note que este sitio es DIFERENTE a los sitios de aplicación del parche de inducción y del parche de desafío.
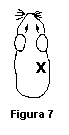
Remover el parche después de 6 horas de aplicación y evaluar a las 24 y 48 horas después de la remoción del parche, como se indica en el punto 6.4.1.3.3, utilizando la escala incluida en la Tabla 2.
Los animales utilizados en este estudio no podrán usarse nuevamente en una prueba de sensibilización.
TABLA 2
ESCALA DE EVALUACION
0 = No hay evidencia de reacción
± = Eritema muy ligero, apenas perceptible, que se presenta sólo en algunas zonas en el área de aplicación del parche
1 = Eritema ligero confluente (reacción ligera pero definida en todo el sitio del parche) o eritema moderado pero irregular (eritema moderado desarrollado en el 50% del área del parche)
2 = Eritema moderado evidente
3 = Eritema severo con o sin edema
6.4.1.10 Análisis de datos
Tabular las evaluaciones como se indica en las tablas incluidas en el apéndice normativo A 1 y 2. Una comparación de los resultados entre los animales del grupo de prueba y de los grupos de control permite la identificación de la sensibilización.
Los resultados se expresan en términos de incidencia y severidad de la respuesta.
Incidencia: El número de animales que muestra una calificación mayor de 1 a las 24 o 48 horas dividido entre el número de animales tratados.
Severidad: La suma de las calificaciones divididas entre el número de animales tratados.
6.4.1.11 Interpretación de los resultados
Como regla general, un material puede clasificarse como un sensibilizante por contacto si se produce una reacción con calificación mayor de 1 en 20% o más en los animales de prueba, siempre y cuando la incidencia en el grupo de control sea menor de la mitad que en el grupo de prueba, además por lo menos en uno de los cobayos del grupo de prueba que presentaron reacción, ésta deberá haber persistido durante 48 horas.
A continuación se incluyen ejemplos que ilustran resultados de prueba de sensibilización y su interpretación.
Ejemplo Grupo Incidencia (*) Interpretación
1 Prueba 4/20 Positiva
Control 0/10
2 Prueba 9/20 Positiva
Control 2/10
3 Prueba 10/20 Cuestionable
Control 3/10
4 Prueba 3/20 Cuestionable
Control 0/10
5 Prueba 0/20 Negativa
Control 1/10
(*) Número total de animales que muestran una reacción de la piel ³ a 1 a las 24 o 48 horas después del tratamiento, con respecto al número total de animales incluidos.
6.4.1.12 Criterio de aceptación
Los productos para los que se obtenga una interpretación NEGATIVA en esta prueba de sensiblización, serán aceptados como productos aptos para uso humano.
Si en un producto se obtiene una interpretación CUESTIONABLE, se podrá optar por cualquiera de las siguientes alternativas:
6.4.1.12.1 Ostentar la leyenda "Este producto puede ser sensibilizante".
6.4.1.12.2 Repetir la prueba. Si los resultados de esta segunda prueba confirman los obtenidos en la primera, deberá optarse por la alternativa del punto 6.4.1.12.1, si la interpretación obtenida en la segunda prueba es negativa, el producto será considerado apto para uso humano.
6.4.1.12.3 Proceder a la prueba en humanos. Si los resultados son negativos, se considerará el producto apto para uso humano.
6.4.2 Prueba de sensibilización en humanos
Esta prueba tiene por objeto proporcionar información con respecto al potencial de sensibilización (desarrollo de reacciones alérgicas) como consecuencia de exposiciones repetidas del material de prueba en piel intacta. Esto se evalúa en voluntarios, mediante contacto externo del material de prueba con la piel, utilizando un procedimiento de aplicaciones repetidas con parche ocluidos.
6.4.2.1 Materiales y reactivos
Agua destilada esterilizada
Fuente de luz blanca mínimo de 60 watts
Lente de aumento
Matraz Erlenmeyer
Gasa quirúrgica esterilizada
Tela adhesiva o micropore
Tela ahulada
Mortero
Balanza analítica
Tamiz del No. 40 (0.42 mm de abertura de malla)
Para desarrollar este estudio se requiere la participación de un mínimo de 80 voluntarios.
6.4.2.2 Procedimiento
Cada uno de los voluntarios, previo a su participación en el estudio, debe proporcionar al investigador un documento en que exprese su consentimiento a participar en esta prueba. Un modelo de este tipo de documento se incluye en el apéndice informativo A. El investigador informará a cada voluntario del propósito y naturaleza del estudio.
6.4.2.3 Elección de los voluntarios
La persona puede ser elegible para este estudio, de acuerdo a los criterios que se mencionan en el punto 6.3.2.1 y además los siguientes:
Si una persona participó anteriormente en una prueba de sensibilización y no se le desarrolló alergia, puede participar nuevamente en una prueba de este tipo, sólo si ya han transcurrido un mínimo de 6 meses de la prueba anterior.
Si una persona participó anteriormente en una prueba de sensibilización y se le desarrolló alergia, no podrá participar nuevamente en un estudio de este tipo.
La elección de cada sujeto potencial se determina a través del llenado de un cuestionario que cubre aspectos de historia alérgica y dermatológica del paciente (ver apéndice informativo A 3). Los sujetos se excluirán de la prueba por una o más de las razones expuestas anteriormente. El centro de prueba podrá, a su criterio, incluir casos adicionales para determinar la elección.
Todas las decisiones con relación a la elección deben ser documentadas adecuadamente haciendo las anotaciones pertinentes en el cuestionario correspondiente a cada sujeto. Cuando sea necesario, el supervisor podrá consultar con un dermatólogo para determinar la elección de un sujeto.
6.4.2.4 Aplicación de los parches
Los sitios de la aplicación de los parches deben ser marcados adecuadamente, de manera que la localización de cada sitio de aplicación de parche quede indicada claramente. Esto puede realizarse utilizando una solución de violeta de genciana al 0,5%. Este marcado de los sitios de aplicación de los parches permite la aplicación en los mismos sitios a lo largo de la fase de inducción del estudio.
En cada parche debe aplicarse 0,3 ml (líquidos) o 0,3 g (sólidos, polvos o pastas) de la sustancia de prueba. La sustancia de prueba debe prepararse de la siguiente manera:
En el caso de productos que el consumidor utiliza al 100% y se dejan aplicados sobre la piel todo el día, tales como cremas, talcos, etc., éstos deben ser aplicados en el parche tal y como se presentan a la venta.
Para productos que se emplean diluidos (champúes (shampoos), jabones, etc.), los que se usan al 100% pero que no se aplican directamente sobre la piel durante su uso normal (máscaras para pestañas, etc.), y en los que se utilizan al 100% y se enjuagan un tiempo después (tintes, ondulantes, fijadores, depilatorios, etc.), la sustancia de prueba se debe preparar como una dilución empleando un vehículo adecuado (agua destilada, aceite mineral, etc.) dependiendo de la naturaleza del producto. En este caso, la concentración de la sustancia de prueba que se aplique en el parche debe ser la que provoque una mínima irritación en la piel humana en una prueba de parche.
Para productos que presentan un solvente (como lociones, quita esmaltes, etc.) éste debe dejarse evaporar durante 15 minutos antes de efectuar la prueba.
Para los productos que se aplican diluidos en la prueba, debe incluirse un parche en que se aplique el vehículo, a modo de control, además del parche con la sustancia de prueba.
Para asegurar una buena adhesión de los parches, éstos se aseguran sobre la piel utilizando tela adhesiva porosa, como micropore o dermicel. Estas "bandas de refuerzos" deben ser aplicadas de manera que ejerzan una presión homogénea sobre el parche.
Los materiales de prueba se aplican a los participantes en una secuencia establecida previamente. Esta secuencia de aplicación debe rotarse entre los participantes con objeto de minimizar sesgos debidos a la posición del parche en el brazo, de la siguiente manera:
Cuatro muestras: 1-2-3-4, 2-3-4-1, 3-4-1-2, 4-1-2-3
Tres muestras: 1-2-3, 2-3-1, 3-1-2
Dos muestras: 1-2, 2-1
Esta secuencia debe ser documentada claramente para cada participante a lo largo de la prueba.
6.4.2.5 Inducción
Los parches con el material de prueba deben ser aplicados en la superficie lateral superior del brazo, en el orden asignado a cada sujeto.
Antes de la aplicación del primer parche de inducción y de la aplicación de los parches de desafío, el área del brazo a tratar debe limpiarse con una gasa saturada con alcohol al 95% (etanol o isopropanol) para remover grasa superficial de la piel y exceso de humedad. Debe permitirse la evaporación del alcohol antes de que se apliquen los parches.
El brazo de inducción se determina por el supervisor de la prueba y se anota. Este brazo no debe tener cicatrices grandes de vacuna, tatuajes, vitiligios o cualquier otro tipo de cicatrices.
Los parches se aplican longitudinal y centralmente entre el hombro y el codo en la superficie lateral del brazo, con el brazo en una posición relajada al lado del cuerpo. Cada parche de inducción se aplica al mismo sitio que el parche previo con la sustancia de prueba, a menos que el grado de reacción a ésta haya sido tal que se necesite reubicación. Los sitios alternativos en estos casos deben estar adyacentes al sitio original ya sea hacia la parte anterior o la posterior de la superficie lateral del brazo. Después de colocar un parche en ambos sitios adyacentes durante la fase de inducción, el parche debe ser colocado nuevamente en la localización original central, siempre y cuando haya desaparecido la respuesta inicial. En caso de que no sea adecuado colocar el parche en este sitio central nuevamente, el parche no se aplica hasta que la reacción residual desaparezca totalmente. Si esto ocurre, debe documentarse adecuadamente.
Si sólo una pequeña porción del sitio de aplicación muestra una respuesta suficientemente intensa como para ameritar la relocalización del parche, el siguiente parche debe ajustarse ligeramente para evitar esta área. Este caso no se considera una relocalización completa.
Cada voluntario participante recibe instrucciones para mantener todos los parches tan secos como sea posible y remover o desechar los parches después de 24 horas. El área donde estaba colocado cada parche debe ser limpiada de la manera acostumbrada después de la remoción, pero sin que esto elimine la marca que identifica los sitios de aplicación de los parches. Las personas participantes pueden bañarse después de quitar el parche. Este tipo de instrucciones deben proporcionarse a cada participante por escrito al comenzar la prueba.
Los parches de inducción se aplican en lunes, miércoles y viernes de las primeras 3 semanas de la prueba (nueve aplicaciones en total), y se valúan 48 horas después de su aplicación. En los participantes que se ausenten una vez durante estas 9 aplicaciones, el último parche de inducción deberá aplicarse el cuarto lunes de la prueba. Esta aplicación deberá evaluarse 48 horas más tarde.
6.4.2.6 Desafío
Los parches de desafío se aplican simultáneamente a ambos brazos (de inducción y desafío) de cada participante, durante 24 horas. En el brazo de inducción los parches son aplicados aproximadamente en los mismos sitios que los parches de inducción. Si la reacción residual de la fase de inducción es severa, el parche de desafío deberá aplicarse hasta que esta reacción desaparezca. El parche de desafío se coloca en el brazo correspondiente, en posición similar a la del parche de inducción. Cualquier modificación debe ser debidamente documentada.
Los parches de desafío se aplican aproximadamente 17 días después de la última aplicación de inducción.
Cualquier sujeto que se ausente más de una vez durante la fase de inducción o durante la semana de desafío, debe ser eliminado de la prueba. El número final de individuos que completaron el estudio deberá incluir solamente a aquellos participantes que hayan tenido 9 parches de inducción y cuyo parche de desafío haya sido evaluado 2 veces.
Todos los voluntarios deben ser informados adecuadamente sobre las fechas en que deben asistir al lugar en que se hará la evaluación de la prueba y de los números telefónicos de emergencia. Se sugiere proporcionarles un calendario en que estén claramente marcadas todas las fechas de asistencia, así como una explicación por escrito de los días de los requerimientos pertinentes. Además, deberá proporcionarse el número telefónico de las personas apropiadas relacionadas con la prueba, de manera que éstas puedan ser localizadas cualquier día a cualquier hora.
6.4.2.7 Evaluación de los sitios de prueba
Para iluminar el área del parche durante cada evaluación debe utilizarse una fuente de luz artificial, de preferencia una lámpara con foco azul incandescente de 100 watts. El brazo del sujeto debe permanecer en posición relajada al lado del cuerpo. Los sitios deben ser evaluados 48 horas después de la aplicación del parche durante la fase de inducción (72 horas si el parche se aplicó en viernes). Durante la fase de desafío los sitios de aplicación del parche deben ser evaluados a las 48 y 96 o bien a las 48 y 72 horas después de la aplicación.
La evaluación de los sitios de parche se hará de acuerdo a la escala de evaluación que se incluye en el punto 5.6.1.
La misma persona debe evaluar los sitios de parche para los sujetos durante todo el estudio.
6.4.2.8 Participantes que no completaron la prueba
Si un sujeto se elimina del estudio, debido a que no asistió a todas las sesiones, esto debe ser debidamente documentado.
En estos casos, debe determinarse la razón por la que el individuo no asistió. Si el sujeto se encuentra preocupado por la respuesta a un parche, si asocia algún problema con su participación en la prueba, le disgusta algún aspecto de la prueba, etc., se deben realizar esfuerzos para comunicarse con dicho sujeto, animarlo a seguir participando en caso de que esto sea pertinente, o bien obtener evaluaciones en los sitios de prueba si se va a retirar del estudio. Se debe ofrecer consulta, diagnosis y tratamientos dermatológicos en aquellos casos que así lo requieran.
Se debe dar un seguimiento adecuado en aquellos casos en que un sujeto muestra señales de una posible sensibilización a la sustancia de prueba, independientemente de que se haya retirado o no del estudio. Sin un sujeto demuestra ser alérgico a una determinada sustancia de prueba y se retira del estudio, deben aplicarse parches de desafío para corroborar esta reacción.
6.4.2.9 Guía para la interpretación de los datos
En aquellas respuestas de la piel en los sitios de desafío que sean más severas a la sustancia de prueba, en comparación con la respuesta durante las aplicaciones de inducción, se considera como posiblemente debidas al desarrollo de hipersensibilidad retardada por contacto (alergia). Además, aquellas respuestas que se incrementan en severidad durante las evaluaciones de desafío de 48 a 96 horas, también serán consideradas como posiblemente debidas a una alergia por contacto, mientras que aquellas que disminuyen en severidad de las 48 a las 96 horas, generalmente son indicativas de irritación primaria.
Si se presentan respuestas persistentes de la piel (por 96 horas o más) con pápulas o edema que se presenten durante la primera semana de inducción, se considera generalmente que esto indica una alergia preexistente. Si se desarrollan reacciones persistentes de este tipo durante la segunda o tercera semana de la fase de inducción, se considera que esto puede indicar desarrollo de reacciones alérgicas durante la prueba.
Las respuestas de la piel que sugieren hipersensibilidad por contacto durante esta prueba pueden realmente resultar de irritación primaria y no de alergia cutánea. En estos casos pueden ser muy útiles pruebas de re-desafío para diferenciar entre reacciones alérgicas y reacciones de irritación.
6.4.2.10 Prueba de re-desafío confirmatoria
Cualquier sustancia de prueba sospechosa de haber inducido sensibilización durante la prueba original puede ser aplicada nuevamente al sujeto en cuestión, así como a sujetos no sensibilizados (control) hasta 3 meses después del desafío original, una vez que se ha obtenido su consentimiento. Los parches se aplican en la superficie lateral superior de ambos brazos. Después de 6 horas los parches se quitan y desechan. Se efectúan por lo menos dos evaluaciones de la respuesta, ya sea a las 48 y 96 horas o bien a las 48 y 72 horas después de la aplicación.
6.4.2.11 Escala de evaluación
0 No hay reacción visible. Esta calificación puede incluir reacciones superficiales de la piel, tales como brillo o resequedad.
1 Reacción eritematosa ligera, color rosado pálido o rosado
1E* Reacción eritematosa ligera, con pápulas y/o edema
2 Reacción eritematosa moderada. Eritema definido rosado a rojo
2E* Reacción eritematosa moderada con edema y/o pápulas
3 Reacción eritematosa marcada. Color rojo brillante
3E* Reacción eritematosa marcada con edema marcado, pápulas y/o algunas vesículas
4 Reacción eritematosa severa con eritema, pápulas, edema y vesículas
5 Reacción bulosa
(S) Reacción presente más allá del área del parche
Nota: Se considera que se presenta eritema, pápulas y vesículas cuando éstas involucran 25% o más del sitio del parche.
E* Si se presentan pápulas, añadir una P a la calificación; si se presentan vesículas, añadir una V a la calificación.
Criterio de relocalización: Cualquier calificación mayor o igual a 2 (incluyendo la adición de V o P) durante la inducción, requiere que el parche se relocalice en la siguiente aplicación.
Calificación doble: Indica que el parche se movió a un sitio adyacente al original. La primera calificación será la correspondiente para el sitio nuevo; el segundo número es la calificación de la reacción residual en el sitio original. Generalmente, una reacción residual se califica y reporta una sola vez después de la relocalización. Si se sospecha sensibilización, la calificación residual del sitio original podrá reportarse más de una vez.
7. Pruebas in vitro
7.1 Prueba de la membrana corioalantoica
7.1.1 Materiales y reactivos
10 huevos frescos fertilizados de gallina Lehmann-Leghorn con un peso entre 50 y 60 g por cada concentración de sustancia empleada
Incubadora con charolas de rotado automático
Fresa dental con rotor de baja velocidad (12000 a 15000 rpm)
Microscopio estéreo de alta magnificación
Solución salina isotónica
7.1.2 Procedimiento
Se ponen a incubar los huevos a 37,5 ± 0,5ºC de temperatura, a una humedad de 62,5 ± 7,5% durante 10 días, girándolos al 5o. día para controlar el crecimiento. El décimo día se abren los huevos, por el extremo ancho donde se localiza la burbuja de aire, empleando una fresa dental con rotor, a la membrana interior del huevo se empapa con una solución salina isotónica para removerla mecánicamente, una vez removida ésta, la membrana corioalantoica está lista para analizar productos.
Se le aplican 0,3 ml de la muestra de prueba, se observa directamente o se emplea un estereomicroscopio para observar hasta la reacción más ligera.
7.1.3 Análisis de los datos
Se deja el producto en contacto con la membrana 5 minutos (300 segundos) y se prueban distintas concentraciones. Se observa si se presentan reacciones y se anotará el índice de acuerdo con la siguiente Tabla si no hay reacción detectable, se calificará al producto como no irritante.
TABLA No. 3
REACCION CALIFICACION
Vasos capilares más inyectados de lo normal 1
Enrojecimiento con los vasos difícilmente distinguibles 2
Enrojecimiento intenso 3
Enrojecimiento intenso con inflamación 4
Hemorragia generalizada 5
Lisis (destrucción de vasos) 7
Coagulación 10
(Desnaturalización protéica con trombosis, decoloración, opacidad y turbidez intra y extravascular)
Se califica cada serie de huevos y se obtiene el promedio.
7.1.4 Interpretación de los resultados
De los promedios obtenidos se obtiene una clasificación así:
INDICE CLASIFICACION
de 0.0 a 0.9 No irritante
de 1.0 a 3.9 Ligeramente irritante
de 4.0 a 8.9 Moderadamente irritante
de 9.0 y mayores Severamente irritante
7.1.5 Criterio de aceptación
Los productos para uso en bebés deben ser no irritantes.
Los demás productos, para permanecer en el mercado, deben obtener una calificación no mayor de 4.
Los productos que tengan una calificación mayor de 4 deben ostentar en etiquetas la leyenda: "Producto irritante".
Los productos con una calificación mayor de 9 no podrán destinarse para uso humano.
7.2 Ensayo en ojo de bovino
7.2.1 Materiales y reactivos.
8 ojos sanos de ternera extraídos de animales recién sacrificados se colocan dentro de los 10 minutos siguientes a la extracción en una solución salina (0,15 M de cloruro de sodio en agua) y se transportan a una temperatura entre 15 y 20°C (por cada producto a investigar) se verifican dentro de la solución salina y se descartan los ojos con daños o anormalidades y el resto se mantiene en solución salina.
Equipo para baño maría
Tamiz del No. 40 (0,42 mm de abertura de malla)
Charola portahuevos de plástico
Mortero con pistilo
Anillos de silicón con diámetro interior de 8 mm y diámetro exterior de 13 mm
Solución salina isotónica
Solución de fluoresceína (2% en solución pH 7,0)
Lente de aumento
Fuente de luz blanca
Balanza analítica
Matraz Erlenmeyer de 1,5 ml
7.2.2 Procedimiento
Los ojos se emplean dentro de las 4 horas siguientes a su extracción, se destinan 5 a la prueba y 3 como controles y se colocan en una atmósfera húmeda en el baño maría cerrado por 5 minutos, se les aplican 0,1 ml del producto de prueba si es líquido y si es sólido se pulveriza en un mortero hasta hacerlo pasar por un tamiz del No. 40 y se calcula la dosis a aplicar mediante una multiplicación de la densidad del producto en mg/ml por 0,1. La cantidad a aplicarse debe pesarse con una precisión del 1% utilizando una balanza analítica.
Se aplica la sustancia de prueba directamente en la córnea dentro del anillo, en los 5 ojos de prueba, de los ojos de control uno se deja sin tratar y a los otros se les aplica 0,1 ml de tolueno y acetona, respectivamente, después de 30 segundos los ojos se riegan con solución salina 10 ml en total, se continúa la incubación en el baño maría cerrado, 10 minutos más; se les aplica fluoresceína y se examinan los resultados.
7.2.3 Interpretación de los resultados
Se examina con una lente de aumento auxiliándose con una fuente de luz, los ojos de prueba, se observa la integridad del epitelio de la córnea, las calificaciones se establecen de acuerdo a la Tabla No. 4, la calificación final se obtiene sumando las calificaciones de opacidad, integridad epitelial y desprendimiento epitelial y dividiendo entre el número de ojos expuestos en cada caso.
TABLA No. 4
OPACIDAD CALIFICACION INTEGRIDAD CALIFICACION DESPRENDIMIENTO CALIFICACION
EPITELIAL EPITELIAL
(GRADO DE TINCION)
NINGUNA 0 NINGUNA 0 SIN GRAVES 0
ANORMALIDADES
LIGERA 1 DIFUSA O DEBIL 0,5 ARRUGAMIENTO DE 2
LA SUPERFICIE
MARCADA 2 CONFLUENTE Y 1 PERDIDA DE 3
DEBIL EPITELIO
SEVERA 3 CONFLUENTE E 1,5 AUSENCIA DE 4
INTENSA EPITELIO
CORNEA OPACA 4
7.2.4 Criterio de aceptación
Todos los productos para uso en bebés deberán obtener resultados de 0 (cero) en los tres parámetros.
Los demás productos deberán obtener una calificación de opacidad no mayor de 2, una de integridad del epitelio no mayor de 0,5 y no mayor de 0 (cero) de desprendimiento epitelial.
Los productos que obtengan calificaciones mayores podrán repetir la prueba o imprimir en etiquetas la nota "Este producto es irritante".
7.3 Ensayo en células rojas de sangre
7.3.1 Material
Células rojas de sangre
Centrífuga clínica estándar (10 000 rpm)
Espectrofotómetro UV visible
Tubo de ensayo
Dodecil sulfato de sodio al 1% ( solución estándar)
Matraces Erlenmeyer suficientes
7.3.2 Procedimiento
7.3.2.1 Preparación de las células rojas
Las células rojas de sangre se aíslan por centrifugación de sangre citrada de ternera o cerdos recién sacrificados y lavada con solución de buffer salino fosfatado isotónico hasta eliminar trazas de plasma y células blancas.
Para uso posterior, cuidadosamente se preparan suspensiones de células rojas de sangre conteniendo glucosa (10 mmol/l) y se ajustaron para rendir aproximadamente 8 x 109 células/ml.
La suspensión se prepara semanalmente y se almacena a 4ºC, en condiciones adecuada puede durar hasta 6 semanas con leve pérdida de 25%.
7.3.2.2 Hemólisis
Se preparan alícuotas de suspensión de células rojas con concentración fija de 0,125 mmol/1 de oxihemoglobina, se incuban por 10 minutos a temperatura ambiente con agitación, asimismo diluciones del dodecil-sulfato de sodio al 1% con incrementos equivalentes de la concentración de la muestra de prueba, para establecer una relación concentración-respuesta.
El periodo de incubación se termina con una corta y rápida centrifugación (1 minuto a 10 000 rpm). El sobrenadante resultante se monitorea fotométricamente para hemólisis a 530 nm y 560 nm contra un blanco y un control.
La experiencia anota que a una concentración de 0,125 mmol/l de dodecil sulfato de sodio la desnaturalización es total (100%).
A partir de este dato se traza una curva de respuesta concentración.
7.3.2.3 Desnaturalización de proteínas
El producto de prueba a una concentración del 1%, se incuba 10 minutos a temperatura ambiente, con una alícuota de células rojas ajustadas a la concentración de oxihemoglobina, previamente descrita. Agitar vigorosamente y centrifugar a 10 000 rpm, el sobrenadante se analiza a 540 y 575 nm en el espectrofotómetro.
La extinción medida a 575 nm se divide por la extinción medida a 540 nm para obtener el llamado Radio (R1) que es usado para calcular el índice de desnaturalización de hemoglobina DI (%). Para la oxihemoglobina, el radio R1 es 1,05 ± 0,001, mientras que el estándar interno (SDS) (a 3,5 mmol/l como concentración final de prueba) da una relación R2. La diferencia R1-R2 se define como igual al 100% de desnaturalización de oxihemoglobina y es usado para calcular la potencia relativa individual de desnaturalización de las muestras, de acuerdo a la siguiente fórmula:
100 (R1-Rs)
DI (%) = ------------------------
(R1-R2)
Donde:
Rs = Radio obtenido por la muestra
7.3.3 Interpretación de los resultados
La inclinación positiva de la curva, muestra el aumento en el daño celular, la liberación inmediata de oxihemoglobina se refleja en el aumento de absorbancia a mayor concentración del estándar al haber destrucción celular las absorbancias a 540 y 575 nm disminuyen por la desnaturalización; la absorbancia a 560 nm no cambia.
A partir de esta curva se hace el ensayo a distintas concentraciones de prueba de la sustancia a investigar con las células rojas y se estima su índice de desnaturalización.
7.3.4 Criterio de aceptación
Los productos para uso humano no deberán presentar ningún índice de desnaturalización de células rojas de sangre.
7.4 Ensayo membrana-reactivo
7.4.1 Materiales y reactivos
Membrana proteica de queratina y colágeno disponible (ya preparada en el comercio especializado)
Reactivo de globulinas y colágeno liofilizado (disponible y preparado en el comercio especializado).
Fotocolorímetro con celdillas suficientes
Colorante rojo básico No. 2
Incubadora (para mantener la temperatura a 25 ºC)
Soporte de (anillo) disco
Estándares de lauril sulfato de sodio a distintas concentraciones
7.4.2 Procedimiento
7.4.2.1 Matrices
Se prepara en una solución salina estabilizada a pH 8,0 de 10% de queratina 1% colágeno, 0,1% glutaraldehído y acetato de celulosa (suficiente); se deja a 25ºC durante una hora, a esta preparación, se le adiciona colorante rojo básico No. 2 y se deja 10 minutos a 25ºC.
Las películas que se forman se lavan exhaustivamente con agua destilada se cortan en círculos y se montan en soportes de disco, se guardan en recipientes de plástico a 4ºC conservándose así hasta 180 días.
7.4.2.2 Reactivo
Un polvo liofilizado de globulinas, colágeno, glicosaminoglicanos, ácidos grasos libres, aminoácidos, fosfolípidos y sales buffer, se rehidrata con agua destilada a 4ºC y se conserva así hasta 15 días.
7.4.2.3 Curva dosis-respuesta
Se determina el índice de irritación primaria dérmica IN VIVO y además con este procedimiento empleando soluciones estándares de lauril sulfato de sodio (u otro irritante conocido) a distintas concentraciones se traza una curva de equivalencia entre los índices IN VIVO e IN VITRO, medido como densidad óptica a 470 nm
7.4.2.4 Aplicación de la muestra
Las muestras se preparan a dosis de 30 µl hasta 100 µl en parches de 0,25 cm3, se aplican directamente en la barrera, luego ésta se inserta en el reactivo y se incuba por 3 horas los sólidos se aplican sin diluir, perfectamente molidos y tamizados.
Se determina la densidad óptica a 470 nm en un fotocolorímetro se extrapola en la curva de dosis respuesta y se lee como el equivalente de la prueba IN VITRO y el índice de irritación primaria dérmica IN VIVO.
7.4.3 Interpretación de los resultados
La clasificación de irritación dérmica IN VITRO corresponde directamente con la clasificación IN VIVO (en piel de conejos) así:
INDICE DE IRRITACION PRIMARIA
Prueba Prueba Calificación Calificación
IN VIVO IN VITRO IN VIVO IN VITRO
0-0,5 0-0,5 No irritante Mínimo Irritante
0,51-1,5 0,51-1,5 Ligeramente Levemente
1,6-3,0 1,6-2,0 Levemente Levemente
2.1-3.0 Levemente Moderadamente
3,1-5,0 3,1-5,0 Moderadamente Moderadamente
5,1 y mayores 5,1 y mayores Severamente Severamente
7.4.4 Criterio de aceptación
Los productos para uso en bebés deberán tener un índice de irritación menor a 0,5
Los demás productos deberán tener un índice no mayor de 3 en esta prueba para considerarse aptos para uso humano o bien deberán incluir en sus etiquetas la leyenda " Este producto es irritante".
Debe anotarse que esta prueba sustituye a los ensayos para determinación de irritación primaria dérmica, pero no puede proporcionar ningún indicio sobre respuestas alérgicas o de velocidad de recuperación después de una reacción por lo que no sustituye a las pruebas de sensibilización.
7.5 Ensayo de desnaturalización de proteínas
7.5.1 Materiales y reactivos
Fotocolorímetro
Filtros a 400 nm, 430 nm, 730 nm
Celdillas de plástico de 1,5 ml (100)
Solución de formacina al 5%, al 10% y al 20% en agua destilada.
Papel pH
Soluciones conocidas de irritantes por ejemplo lauril sulfato de sodio al 5%, 10%, 15%, 20% y 25%
Reactivo proteico en polvo para prueba de desnaturalización IN VITRO (disponible en el mercado especializado).
Activador del reactivo (disponible en el mercado)
Micropipetas de 100 µl
Tapas de plástico para las celdillas
Dosificador Eppendor + con micropipetas de 250 µl a 1 ml con incrementos de 250 µl
Micropipetas de 20 a 200 µl
Soportes para las celdillas
Incubadora
Papel filtro grande de 8 pulgadas de diámetro
Embudo de gran capacidad
Probeta graduada
Matraz Erlenmeyer con tapón
Ampolletas reutilizables
Espátulas
Pipeta de rocío
7.5.2 Procedimiento
7.5.2.1 Unidades de medición
Para fines prácticos de medición del grado de desnaturalización se emplean unidades de densidad óptica multiplicadas por 1000 que se reportan como "unidades de opacidad".
La densidad óptica de las soluciones de formacina que se emplean como calibradores deberá estar en los siguientes intervalos:
Solución Densidad óptica Unidades de opacidad
5% 0,400 - 0,650 400 - 650
10% 0,800 - 1,050 800 -1050
20% 1,350 - 1,800 1350 -1800
7.5.2.2 Determinación de la densidad óptica
Para determinar si este es el procedimiento adecuado, se deberá desarrollar lo siguiente: se colocan 100 ml o 100 mg de la muestra de prueba en 1 ml de disolvente en una celdilla. Se ajusta el colorímetro a cero con una celdilla que contenga sólo disolvente, después se lee la muestra a 400 nm si la densida óptica es mayor de 0,5 se deberá optar por la técnica de membrana, si es menor se puede emplear esta técnica.
7.5.2.3 Prueba de pH
Se determinará el pH de la muestra, si éste es mayor de 8,5 deberán aplicarse modificaciones a la técnica.
7.5.2.4 Control de calidad
Para todos los casos realice pruebas con controles conocidos cada 60 días por triplicado en ensayos separados para verificar procedimientos.
7.5.3 Curva dosis-respuesta
Se determina el índice de irritación primaria dérmica IN VIVO y además con esta técnica, empleando soluciones estándar de un irritante conocido a diferentes concentraciones se traza una curva de equivalencia entre los índices IN VIVO y las IN VITRO medidas como densidad óptica a 400 nm o a la que corresponda en muestras coloridas y reactivos.
7.5.4 Cálculo de la densidad óptica
Prepare una solución del reactivo proteico, añadiendo agua destilada hasta dilución total, filtre y transfiera el reactivo empleando una pieza de papel filtro ajustada al embudo y una ampolleta para reactivo, deje que se filtre lentamente (de 30 a 45 minutos) etiquete con la fecha de elaboración. El reactivo es estable por 30 días almacenado de 4º a 8ºC.
Para activar el reactivo añada el activador en proporciones IX, 3X a 5X así, si el pH es menor de 8,5 adicione: IX o sea 0,5 ml de activador a 25 ml del reactivo preparado (la activación se efectúa antes de cada ensayo), si se va hacer en proporción 3X se adicionan 1,5 ml de activador a 25 ml de reactivo y si es 5X, 2,5 ml de activador a 25 ml de reactivo si el pH es mayor de 8,5 no active el reactivo.
Se preparan soluciones de la muestra a partir de 1000 mg/kg
Se aplican a diferentes concentraciones del reactivo, y se determina la densidad óptica del reactivo desnaturalizado a 400 nm, o a la que corresponda según el tipo de muestra y se extrapola en la curva de dosis respuesta.
7.5.5 Muestras que no califican
Si la muestra es muy colorida dará lecturas demasiado altas, entonces deberá cambiarse el filtro para tener una lectura a 470 nm o 730 nm
Si la muestra es reactiva y afecta la celdilla se analiza poniendo primero el reactivo o el disolvente y añadiendo la muestra. Si todavía reacciona el producto puede medirse en tubos de ensayo, si sigue habiendo reacción o vaporización debe optarse por otro método.
7.5.6 Interpretación de los resultados
La clasificación de irritación IN VITRO corresponde con la IN VIVO extrapolada de la curva y debe leerse así:
I N D I C E CLASIFICACION
0 - 0,5 No irritante
0,51- 1,5 Ligeramente irritante
1,6 - 3,0 Moderadamente irritante
3,1 - 5,0 Irritante
5,1 - mayores Severamente irritante.
7.5.7 Criterios de aceptación
Los productos para uso en bebés deberán ser no irritantes. Los demás productos deberán tener índice no mayor de 3 en esta prueba para considerarse aptos para uso humano, o bien deberán incluir en sus etiquetas la leyenda "Este producto es irritante".
8. Muestreo
El procedimiento de muestreo para los productos objeto de esta Norma, se sujetará a lo que establece la Ley General de Salud.
9. Concordancia con normas internacionales
Esta Norma no tiene concordancia con normas internacionales
10. Bibliografía
10.1 Secretaría de Comercio y Fomento Industrial.1992 Ley sobre Metrología y Normalización. México, D.F.
10.2 Secretaría de Salud.1984 Ley General de Salud. México, D.F.
10.3 Secretaría de Salud. 1988. Reglamento de la Ley General de Salud en Materia de Control Sanitario de Actividades, Establecimientos, Productos y Servicios. México, D.F.
10.4 Borenfreund E and Borrero O. 1984. In vitro citotoxicity assays. Potential alternatives to the Draize ocular irritancy test. Cell biol. toxicol. 1,55.
10.5 Golberg, A.M. 1989. In alternative methods in toxicology. Vol. 7 In vitro toxicology. New Directions. (ed) Mary Ann Liebert Inc. New York. pp 297-305. USA.
10.6 Gordon V.C. and Bergman, H.C. 1987,1988,1989, Eytex An In Vitro Method for Evaluation of Ocular Irritancy, Progress in In Vitro Toxicology, Progress in In Vitro Toxicology, Approaches to Validation, Goldberg, A.M.,ed. Mary Ann Liebert, Inc N.Y., pag 1-13, N.Y. USA.
10.7 Griffith F. John, Nixon A. Gerald, Bruce D. Robert, Reer J. Paul, and Bannan A. Elmer. 1980. Dose-Response Studies with Chemical Irritants in the Albino Rabbit Eye as a Basis for Selecting Optimum Testing Conditions for Predicting Hazard to the Human eye, Academy Press, Inc., pag. 501-513. Cincinnatti Ohio. USA.
10.8 Klecak Georg. Identification of Contact Allergens Predictive Test in Animals, Departament of Pharmaceutical Research F. Hoffmann, Chapter 9 pag. 305-328 Switzerland.
10.9 Magnusson Bertil, Kligman M. A. 1969. Usefulness of Guinea Pig Tests for Detection of Contact Sensitizers, Departament of Dermatology University of Lund Malmo, Pennsylvania, School of Medicine Philadelphia, Chapter 23, pag. 551-559. USA.
10.10 Marzulli F.N. and Maibach H.I. 1975. The Rabbit As a Model for Evaluating Skin Irritant:A Conparison of Results Obtained on animals and Man Using Repeated Skin Exposures, Maibch University or California, San Francisco Medical Center pags. 533-540 San Francisco California. USA.
10.11 Mc.Creesh H. Arthur and Steinberg Marshall. Skin Irritation Testing in Animals, Army Enviromental Hygiene Agency Inc, Chapter 5, pag. 193-209.
10.12 Nixon G.A., Bannan C.A., Gaynor T.W., Johnston D.H. and Griffith J.F. 1990. Evaluation of Modified Methods for Determining Skin Irrritation, Regulatory Toxicology and Pharmacology, pag. 12,127-136, Cincinnatti Ohio. USA.
10.13 Rempe Jennifer.1990. Comments on Mexican Board of Health Skin Irritation Testing Protocols, International Affairs Analyst, pag.1-7
10.14 Robinson K. M., Danneman and Nusair L. Terresa. 1989. A Risk Assessment Process for Allergic Contact Sensitization. Fd. Chem. Toxic Vol. 27 No.7 pags. 479-489. Cincinnatti, Ohio. USA.
10.15 Robinson K. Michael, Nusair L. Terresa, Fletcher E. Robert and Ritz L. Harry. 1990. A Review of the Buehler guinea pig skin sensitization test and its use in a risk Assesssment Process for Human Skin Sensitization. Elsevier Scientific Publishers Ireland Ltd, pag. 91-107, Cincinnatti, Ohio. USA.
10.16 Secretaría de Comercio y Fomento Industrial. Norma Oficial Mexicana NOM-008-SCFI-1993. Sistema General de Medida. México, D.F.
10.17 Shelanski, H.A. and Shelanski, M.V. 1983. A new technique of human patch tests. Proc. Sci. Sect. Toilet Goods Assoc., pag 19-46. USA.
10.18 Stotts Jane. 1980. Current concepts in cutaneous toxicity. Planning, conduct and interpretation human predictive sensitization patch tests. Academy Press, Inc. pag 41-53. Cincinnati, Ohio. USA.
10.19 Weterings J.J.M. Peter and Van Erp H.M. Yvette, Validation of the Becamm Assay- An Eye Irritancy Screening Test, Toxicological Research and Consultancy C.V. pags. 515-521.
10.20 Wolfgang J.W. Pape, Ado Hoppe. 1991. In Vitro Methods for the Assessment of Primary Local Effects of topically Applied Preparations, Skin Pharmacology, Editor H. Schroter, Valbonne, pag. 205-213. Hamburgo. Alemania.
10.21 Wolfgang J.W. Pape,V. Pfannenbeckr, U. Hoppe. 1987. Validation of the Red Blood Cell Test System, As in Assay for the Rapid Screeninig of Surfactants, 1987, Molecular Toxicology Publishing Corporation, pag. 525-535. Hamburgo, Alemania.
10.22 Wolfgang J.W. Pape. 1989. In Vitro Toxicity Testing of Surfactants and Tensioactive Cosmetic Products, Beiersdorf A.G. Hamburg, pag. 1-14. Alemania.
11. Observancia de la Norma
La vigilancia del cumplimiento de la presente Norma corresponde a la Secretaría de Salud.
12. Vigencia
La presente Norma Oficial Mexicana entrará en vigor con su carácter obligatorio a los treinta días siguientes a partir de su publicación en el Diario Oficial de la Federación.
Sufragio Efectivo. No Reelección.
México, D.F., a 29 de noviembre de 1994.- El Director General de Control Sanitario de Bienes y Servicios, José Meljem Moctezuma.- Rúbrica.
Apéndice normativo A
A REPORTES
1 Reporte de Incidencia
Concentración de Inducción:
Concentración de Desafío:
Desafío
Grupo de Prueba Grupo Control
Calificación 24 Horas 48 Horas 24 Horas 48 Horas
0
+
1
2
3
(A)
(B)
Total (*)
(A) Número de animales incluidos
(B) Número de animales con calificación 1
(*) El "total" es el número máximo de animales con calificación 1 a las 24 o a las 48 horas
2 Reporte de Incidencia
Concentración de Inducción:
Concentración de Desafío:
Concentración de Re-desafío:
Re-desafío
Grupo de Prueba Grupo Control
Calificación 24 Horas 48 Horas 24 Horas 48 Horas
0
+
1
2
3
(A)
(B)
Total (*)
(A) Número de animales incluidos
(B) Número de animales con calificación 1
(*) El "total" es el número máximo de animales con calificación 1 a las 24 horas o las 48 horas.
Apéndice Informativo A
A FORMATOS
1 CARTA DE CONSENTIMIENTO
Yo _____________________ en pleno uso de mis facultades mentales estoy de acuerdo en usar el producto _____________________________________ en forma voluntaria y sin presión alguna, pudiendo desistir del estudio sin mayor aviso.
También señalo que he sido advertido de los posibles efectos adversos del producto en cuestión.
_________________________ _________________________
F E C H A F I R M A
2 CUESTIONARIO PARA LA PRUEBA DE IRRITACION EN PIEL EN HUMANOS
Participante No. ________
Nombre: _____________________________________________ Edad__________ Sexo (M/F)________
Por favor conteste "sí" o "no" a cada una de las siguientes preguntas:
1. Ha padecido alguna vez:
No Sí ¿Actualmente Describa las la padece? áreas afectadas
Psoriasis ____ ____ ___________ _______________
Eczema ____ ____ ___________ _______________
Otros problemas
de la piel ____ ____ ___________ _______________
Asma ____ ____ ___________ _______________
Enfermedad del heno ____ ____ ___________ _______________
Rinitis alérgica ____ ____ ___________ _______________
2. ¿Padece ud. asma severo? Sí _____ No _____
3. ¿Ha padecido alguna vez cáncer en la piel? Sí ____ No ____
¿Padece esta enfermedad actualmente? Sí ____ No ____
4. ¿Ha visitado alguna vez a su médico con relación a alergias o a algún problema específico en la piel? Sí ____ No ____ ¿Lo padece actualmente? ____ ¿Cual fue/es su problema?
5. ¿Se encuentra usted usando algún medicamento con esteroides (administrado oralmente o aplicado como crema) en el tratamiento de alergia por contacto o erupciones en la piel? Sí__ No __
6. ¿Se le están administrando inyecciones de insulina para el tratamiento de diabetes? Sí __ No___
7. ¿Ha tenido usted algún transplante de órgano que requiera el uso de medicamentos inmunodepresores? Sí____ No ____
8. ¿Está usted recibiendo tratamiento para cualquier tipo de cáncer? Sí ____ No _____
9. Padece usted algún tipo de enfermedad de inmunodeficiencia (lupus, tiroiditis, etc.) Sí __ No __
10. ¿Ha participado usted alguna vez en una prueba de parche? Sí ____ No ____ ¿Cuándo?______________ ¿Dónde?________________
11. ¿Es usted alérgico a alguno de los siguientes productos?
Sí No Mencione el nombre del producto
Shampoos ____ ____ __________________________
Jabones de tocador ____ ____ __________________________
Cremas de belleza ____ ____ __________________________
Perfumes ____ ____ __________________________
Lociones ____ ____ __________________________
Desodorantes/antiperspirantes ____ ____ __________________________
Sombras para los ojos ____ ____ __________________________
Bronceadores ____ ____ __________________________
Depiladores ____ ____ __________________________
Otros productos
cosméticos ____ ____ __________________________
12. ¿Se le han extirpado nódulos linfáticos axilares? Sí____ No _____
13. ¿Se le ha practicado mastectomía? Sí, lado derecho _____, Sí, lado izquierdo ____, Sí, ambos lados ____ No _____
14. Para ser contestada solamente para MUJERES:
¿Está usted tomando actualmente pastillas anticonceptivas? Sí____ No _____
¿Está usted esperando bebé? Sí ____ No ____ ¿Está usted en periodo de lactancia? Sí __No __
Tiene usted antecedentes de trastornos mentales? Sí ____ No ____
Firma ________________________ Fecha ______________________
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
PARA SER LLENADO EXCLUSIVAMENTE POR EL EVALUADOR DEL CUESTIONARIO
1. ¿Esta persona es elegible para participar en la prueba?
Sí ____ No____
Razones
2. Comentarios
Nombre del evaluador____________________________________Firma______________________
Fecha__________________
3 CUESTIONARIO PARA LA PRUEBA DE SENSIBILIZACION EN PIEL EN HUMANOS
Participante No. ________
Nombre:____________________________ Edad______ Sexo (M/F)___
Por favor conteste "sí" o "no" a cada una de las siguientes preguntas:
1. Ha padecido alguna vez:
No Sí ¿Actualmente Describa las
la padece? áreas afectadas
Psoriasis ____ ____ ___________ _______________
Eczema ____ ____ ___________ _______________
Otros problemas
de la piel ____ ____ ___________ _______________
2. ¿Padece ud. asma severo? Sí _____ No _____
3. ¿Ha padecido alguna vez cáncer en la piel? Sí ____ No ____
¿Padece esta enfermedad actualmente? Sí ____ No ____
4. ¿Ha visitado alguna vez a su médico con relación a alergias o a algún problema específico en la piel? Sí ____ No ____ ¿Lo padece actualmente? ____ ¿Cuál fue/es su problema?
5. ¿Se encuentra usted usando algún medicamento con esteroides (administrado oralmente o aplicado como crema) en el tratamiento de alergia por contacto o erupciones en la piel? Sí____ No ____
6. ¿Se le están administrando inyecciones de insulina para el tratamiento de diabetes? Sí __ No___
7. ¿Ha tenido usted algún transplante de órgano que requiera el uso de medicamentos inmunodepresores? Sí____ No ____
8. ¿Está usted recibiendo tratamiento para cualquier tipo de cáncer? Sí ____ No _____
9. ¿Padece usted algún tipo de enfermedad de inmunodeficiencia (lupus, tiroiditis, etc.) Sí __ No___
10. ¿Ha participado usted alguna vez en una prueba de parche? Sí ____ No ____ ¿Cuándo?______________ ¿Dónde?________________
11. ¿Es usted alérgico a alguno de los siguientes productos?
Sí No Mencione el nombre del producto
Shampoos ____ ____ __________________________
Jabones de tocador ____ ____ __________________________
Cremas de belleza ____ ____ __________________________
Perfumes ____ ____ __________________________
Lociones ____ ____ __________________________
Desodorantes/antiperspirantes ____ ____ __________________________
Sombras para los ojos ____ ____ __________________________
Bronceadores ____ ____ __________________________
Depiladores ____ ____ __________________________
Otros productos
cosméticos ____ ____ __________________________
12. ¿Se le han extirpado nódulos linfáticos axilares? Sí____ No _____
13. ¿Se le ha practicado mastectomía? Sí, lado derecho _____, Sí, lado izquierdo ____, Sí, ambos lados ____ No ____
14. ¿Para ser contestada solamente para MUJERES:
¿Está usted tomando actualmente pastillas anticonceptivas? Sí____ No _____
¿Está usted esperando bebé? Sí ____ No ____ ¿Está usted en periodo de lactancia? Sí __ No __
¿Tiene usted antecedentes de trastornos mentales? Sí ____ No ____
Firma ________________________ Fecha ______________________
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
PARA SER LLENADO EXCLUSIVAMENTE POR EL EVALUADOR DEL CUESTIONARIO
1. ¿Esta persona es elegible para participar en la prueba?
Sí ____ No____
Razones
2. Comentarios
Nombre del evaluador____________________________________Firma______________________
Fecha__________________