NORMA Oficial Mexicana NOM-059-SSA1-1993, Buenas prácticas de fabricación para establecimientos de la industria químico farmacéutica dedicados a la fabricación de medicamentos.
Al margen un sello con el Escudo Nacional, que dice: Estados Unidos Mexicanos.- Secretaría de Salud.
NORMA OFICIAL MEXICANA NOM-059-SSA1-1993, BUENAS PRACTICAS DE FABRICACION PARA ESTABLECIMIENTOS DE LA INDUSTRIA QUIMICO FARMACEUTICA DEDICADOS A LA FABRICACION DE MEDICAMENTOS.
FRANCISCO J. HIGUERA RAMIREZ, Director General de Insumos para la Salud, por acuerdo del Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, con fundamento en lo señalado por los artículos 39 de la Ley Orgánica de la Administración Pública Federal; 13 Apartado A fracción I, 194, 194 Bis, 195, 197, 201, 210, 212, 213, 214, 257, 258, 259, 260, 261 y demás aplicables de la Ley General de Salud; 3o. fracción XI, 38 fracción II, 40 fracciones I, V, XI y XII, 41, 43, 47 y 52 de la Ley Federal sobre Metrología y Normalización; 9o., 10, 11, 15, 100, 102, 109, 111 y demás aplicables del Reglamento de Insumos para la Salud; 20 fracción II del Reglamento Interior de la Secretaría de Salud, y
CONSIDERANDO
Que con fecha 14 de diciembre de 1994, en cumplimiento de lo previsto en el artículo 46 fracción I de la Ley Federal sobre Metrología y Normalización, la Dirección General de Insumos para la Salud presentó al Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, el anteproyecto de la presente Norma Oficial Mexicana.
Que con fecha 24 de noviembre de 1995, en cumplimiento del acuerdo del Comité y de lo previsto en el artículo 47 fracción I de la Ley Federal sobre Metrología y Normalización, se publicó en el Diario Oficial de la Federación el proyecto de la presente Norma Oficial Mexicana, a efecto de que dentro de los siguientes noventa días naturales posteriores a dicha publicación, los interesados presentaran sus comentarios al Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario.
Las respuestas a los comentarios recibidos por el mencionado Comité, fueron publicadas previamente
a la expedición de esta Norma en el Diario Oficial de la Federación, en los términos del artículo 47
fracción III de la Ley Federal sobre Metrología y Normalización.
Que en atención a las anteriores consideraciones, contando con la aprobación del Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, se expide la siguiente:
NORMA OFICIAL MEXICANA NOM-059-SSA1-1993, BUENAS PRACTICAS DE FABRICACION
PARA ESTABLECIMIENTOS DE LA INDUSTRIA QUIMICO FARMACEUTICA DEDICADOS
A LA FABRICACION DE MEDICAMENTOS.
PREFACIO
En la elaboración de la presente Norma Oficial Mexicana participaron las siguientes Instituciones y Organismos:
SECRETARIA DE SALUD
Dirección General de Insumos para la Salud.
ASOCIACION FARMACEUTICA MEXICANA, A.C.
CAMARA NACIONAL DE LA INDUSTRIA FARMACEUTICA (CANIFARMA)
COLEGIO NACIONAL DE QUIMICOS FARMACEUTICOS BIOLOGOS, MEXICO, A.C.
COMISION INSTITUCIONAL DE BUENAS PRACTICAS DE FABRICACION (CIPAM)
PRODUCCION QUIMICO FARMACEUTICA, A.C.
UNIVERSIDAD NACIONAL AUTONOMA DE MEXICO
Facultad de Química
INDICE
0. Introducción
1. Objetivo y campo de aplicación
2. Referencias
3. Definiciones
4. Símbolos y abreviaturas
5. Organización de un establecimiento
6. Personal
7. Documentación legal y técnica
8. Diseño y construcción de un establecimiento de la industria químico farmacéutica
9. Control de la fabricación
10. Equipo de fabricación
11. Destrucción y disposición final de residuos
12. Concordancia con normas internacionales y mexicanas
13. Bibliografía
14. Observancia de la norma
15. Anexos
I. Clasificación de áreas
16. Vigencia
Introducción
La salud es un factor de suma importancia para el bienestar y desarrollo social de la comunidad, por lo que corresponde al Ejecutivo Federal a través de la Secretaría de Salud, establecer los requisitos que se deben cumplir durante el proceso de fabricación de los medicamentos que garantice la calidad de los mismos.
La Secretaría de Salud ejercerá el control sanitario de los establecimientos, empleando como marco de referencia la presente Norma Oficial Mexicana.
1. Objetivo y campo de aplicación
Esta Norma Oficial Mexicana establece los requisitos mínimos necesarios para el proceso de los medicamentos y/o productos biológicos comercializados en el país, con el objeto de proporcionar medicamentos de calidad al consumidor.
Es de observancia obligatoria en establecimientos de la industria químico-farmacéutica dedicados a la fabricación de medicamentos y productos biológicos para uso humano.
2. Referencias
Para la correcta aplicación de la presente Norma, se sugiere consultar la siguiente norma oficial mexicana:
NOM-028-STPS-1994 Servicios Generales
3. Definiciones
3.1 Acabado sanitario. Terminación que se le da a las superficies interiores de las áreas con la finalidad de evitar la acumulación de partículas viables y no viables y facilitar su limpieza.
3.2 Acondicionamiento. Son las operaciones por las que un producto a granel tiene que pasar para llegar a ser un producto terminado.
3.3 Agua residual de la industria farmacéutica. Agua descargada resultante de las actividades relacionadas con la fabricación de medicamentos.
3.4 Area. Cuarto o conjunto de cuartos y espacios diseñados y construidos bajo especificaciones definidas.
3.5 Area aséptica. Zona comprendida dentro de una área limpia, diseñada y construida para minimizar la contaminación por partículas viables y no viables, manteniéndola dentro de límites preestablecidos.
3.6 Area crítica aséptica. Zona dentro del área aséptica en la cual el producto, los recipientes y/o los dispositivos de cierre esterilizados, están expuestos al medio ambiente.
3.7 Area limpia. Area diseñada, construida y mantenida con el objeto de tener dentro de límites el número de partículas viables y no viables en superficies y medio ambiente.
3.8 Aseguramiento de calidad. Conjunto de actividades planeadas y sistemáticas que lleva a cabo una empresa, con el objeto de brindar la confianza, de que un producto o servicio cumple con los requisitos de calidad especificados.
3.9 Biocarga. Concentración de UFC presentes en un elemento determinado.
3.10 Bioterio. Area especializada en el mantenimiento, control y/o reproducción de diversas especies de animales destinadas para la realización de pruebas de laboratorio.
3.11 Buenas prácticas de fabricación. Conjunto de lineamientos y actividades relacionadas entre sí, destinadas a garantizar que los productos farmacéuticos elaborados tengan y mantengan la identidad, pureza, concentración, potencia e inocuidad, requeridas para su uso.
3.12 Calidad. Cumplimiento de especificaciones establecidas para garantizar la aptitud de uso.
3.13 Calificación. Evaluación de las características de los elementos del proceso.
3.14 Calibración. Conjunto de operaciones que determinan, bajo condiciones especificadas, la relación entre los valores indicados por un instrumento o sistema de medición, o los valores representados por una medición material y los valores conocidos correspondientes a un patrón de referencia.
3.15 Concentración. Cantidad del fármaco presente en el medicamento expresada como peso/peso, peso/volumen o unidad de dosis/volumen.
3.16 Contaminación. Presencia de entidades físicas, químicas o biológicas indeseables.
3.17 Contaminación cruzada. Presencia de entidades físicas, químicas o biológicas indeseables, procedentes de otros procesos de fabricación.
3.18 Retencion temporal. Los productos, materias primas o materiales de acondicionamiento se retienen temporalmente, con el fin de verificar si se encuentran dentro de las especificaciones de calidad establecidas y la regulación correspondiente.
3.19 Documento maestro. Documento autorizado que contiene la información para controlar las operaciones, proceso y actividades relacionadas con la fabricación de un producto.
3.20 Envase primario. Es aquel que contiene un fármaco o preparado farmacéutico y que está en contacto directo con él.
3.21 Envase secundario. Envase dentro del cual se coloca el envase primario.
3.22 Especificación. Descripción de un material, sustancia o producto, que incluye los parámetros de calidad, sus límites de aceptación y la referencia de los métodos a utilizar para su determinación.
3.23 Etiqueta. Cualquier marbete, rótulo, marca o imagen gráfica escrita, impresa, estarcida, marcada, marcada en relieve o en hueco grabado, adherido o precintado en cualquier material susceptible a contener el medicamento incluyendo el envase mismo, en caracteres legibles e indelebles.
3.24 Expediente legal. Conjunto de documentos que demuestran que el medicamento está registrado y cumple con las normas vigentes de la Secretaría de Salud.
3.25 Expediente maestro. Conjunto de documentos que proporcionan la información necesaria para la fabricación de un medicamento.
3.26 Fabricación. Operaciones involucradas en la producción de un medicamento desde la recepción de materiales hasta su liberación como producto terminado.
3.27 Fármaco. Sustancia natural o sintética que tenga alguna actividad farmacológica y que se identifique por sus propiedades físicas, químicas o acciones biológicas, que no se presenten en forma farmacéutica y que reúna condiciones para ser empleada como medicamento o ingrediente de un medicamento.
3.28 Inactivación. Acción de transformar la actividad química/biológica de los residuos medicamentosos inutilizándolos para su uso farmacéutico.
3.29 Lote. Cantidad de un fármaco o medicamento, que se produce en un ciclo de fabricación y cuya característica esencial es su homogeneidad.
3.30 Materia prima. Sustancia de cualquier origen que se use para la elaboración de medicamentos
o fármacos.
3.31 Medicamento. Toda sustancia o mezcla de sustancias de origen natural o sintético que tenga efecto terapéutico, preventivo o rehabilitatorio, que se presente en forma farmacéutica y se identifique como tal por su actividad farmacológica, características físicas, químicas y biológicas.
3.32 Número de lote. Combinación alfanumérica que identifica específicamente un lote.
3.33 Orden de producción. Copia de la fórmula maestra de producción a la cual se le asigna un número de lote y se utiliza como guía y registro de las operaciones para la producción de un lote de medicamento.
3.34 Orden de acondicionamiento. Copia de la fórmula maestra de acondicionamiento a la cual se le asigna un número de lote y se utiliza como guía y registro de las operaciones para el acondicionamiento de un lote de medicamento.
3.35 Partículas viables. Cualquier partícula que bajo condiciones ambientales apropiadas puede reproducirse.
3.36 Principio activo. Ver fármaco.
3.37 Procedimiento normalizado de operación. Documento que contiene las instrucciones necesarias para llevar a cabo de manera reproducible una operación.
3.38 Producto. Es el resultado de un proceso específico.
3.39 Producto a granel. Producto que ha cubierto todas las etapas del proceso de producción y que será sometido a etapas posteriores de acondicionamiento antes de convertirse en producto terminado.
3.40 Producto intermedio. Material parcialmente procesado que será sometido a etapas posteriores de producción antes de convertirse en producto a granel.
3.41 Producto terminado. Medicamento en su presentación final.
3.42 Pureza. Grado en el cual las materias primas, los productos intermedios y a granel, están exentos de materiales extraños.
3.43 Rastreabilidad. Capacidad de reconstruir la historia, localización de un elemento o de una actividad, por medio de registros de identificación.
3.44 Rendimiento final. Cantidad de producto terminado obtenido al final del proceso de fabricación.
3.45 Rendimiento teórico. Cantidad de producto que será obtenida a través de un proceso.
3.46 Sistemas críticos. Son aquellos que tienen impacto directo en los procesos y/o productos.
3.47 Surtido. Entrega de materias primas, producto intermedio, producto a granel y/o materiales.
3.48 Retiro de producto farmacéutico. Acción de recoger un producto del mercado.
3.49 Validación. Es la evidencia documentada que demuestra que a través de un proceso específico se obtiene un producto que cumple consistentemente con las especificaciones y los atributos de calidad establecidos.
4. Símbolos y abreviaturas
Cuando en esta Norma se haga referencia a las siguientes abreviaturas, se entenderá:
BPF buenas prácticas de fabricación.
DGIS Dirección General de Insumos para la Salud.
FEUM Farmacopea de los Estados Unidos Mexicanos.
LNSP Laboratorio Nacional de Salud Pública.
NOM Norma Oficial Mexicana.
LGS Ley General de Salud.
OMS Organización Mundial de la Salud.
PNO Procedimiento Normalizado de Operación.
SEMARNAP Secretaría de Medio Ambiente, Recursos Naturales y Pesca.
SSA Secretaría de Salud.
UFC Unidades Formadoras de Colonias.
5. Organización de un establecimiento
5.1 El establecimiento debe contar con una organización interna acorde con el tamaño de la empresa y los productos que fabrica.
5.2 Debe existir un organigrama detallado y actualizado en donde se identifique claramente que el encargado de producción y el del área de calidad no reporten el uno al otro.
5.3 El responsable sanitario debe ocupar el mayor nivel jerárquico del área técnica o reportar directamente a esta posición o al puesto más alto del establecimiento.
5.4 Debe existir un número suficiente de auxiliares de responsable y supervisores de área para cubrir y supervisar las funciones operativas dentro del horario de trabajo.
5.5 Los encargados de las áreas de producción y calidad, deben tener como mínimo estudios de licenciatura en el área farmacéutica o química, así como título y cédula profesionales.
5.6 El encargado del área de producción se encargará de realizar las siguientes funciones, sin perjuicio de las obligaciones y responsabilidades que correspondan al responsable sanitario, conforme a la Ley General de Salud y al Reglamento de Insumos para la Salud:
5.6.1 Que los productos se fabriquen dentro de especificaciones, de acuerdo con las buenas prácticas de fabricación, procedimientos normalizados de operación y documentos autorizados.
5.6.2 Que las áreas, equipos y sistemas críticos cumplan con lo indicado en la presente Norma.
5.6.3 Que se lleven a cabo estudios de validación de los procesos de fabricación y de los sistemas involucrados.
5.7 El encargado del área de calidad se encargará de realizar las siguientes funciones, sin perjuicio de las obligaciones y responsabilidades que correspondan al responsable sanitario, conforme a la Ley General de Salud y al Reglamento de Insumos para la Salud:
5.7.1 Las aprobaciones y rechazos de todos los componentes utilizados en la fabricación de los productos terminados.
5.7.2 Que todos los análisis se realicen conforme a las buenas prácticas de laboratorio.
5.7.3 Que se cumplan con todos los PNO´s relacionados a la función de calidad.
5.7.4 Que se lleven a cabo estudios de validación de los procesos de fabricación y de los sistemas involucrados.
5.7.5 La asignación de fechas de reanálisis a las materias primas y fechas de caducidad a los productos y reactivos.
5.7.6. Que la documentación relativa a la fabricación y control de los lotes producidos se conserve.
5.7.7 Que por cada queja se realicen las investigaciones correspondientes y asegurarse de que se implementen las acciones correctivas necesarias, si procede.
5.7.8 La evaluación de proveedores.
6. Personal
6.1 Las obligaciones y responsabilidades del personal deben establecerse por escrito.
6.2 Debe existir un programa documentado para la capacitación y entrenamiento del personal en las funciones que le sean asignadas y en lo referente a PNO´s. Este programa debe indicar como mínimo: contenido, participantes, frecuencia y constancia de realización.
6.3 El personal debe portar ropa limpia y confortable y el equipo de protección, diseñado para evitar la contaminación de los productos y de las áreas de fabricación, así como riesgos de salud ocupacional.
6.3.1 Los requerimientos de indumentaria para cada área de fabricación deben estar definidos por escrito.
6.3.2 Se debe contar con un PNO de lavado de indumentaria.
6.3.3. En caso de usar indumentaria desechable se debe contar con un PNO para su disposición final.
6.4 El personal de nuevo ingreso debe pasar un examen médico.
6.5 Se debe hacer periódicamente un examen médico a todo el personal de las áreas de fabricación, así como después de una ausencia debida a enfermedades transmisibles y tomar las acciones necesarias en caso de diagnóstico positivo.
6.6 Debe evitarse la entrada a las áreas de fabricación al personal que padezca infecciones, enfermedad contagiosa o lesiones abiertas.
6.7 Si el personal tiene que salir de la planta, debe cambiarse la ropa de trabajo para volvérsela a poner al momento de reingresar al área de fabricación correspondiente.
6.8 El personal debe cumplir con los PNO´s para cada área de fabricación.
6.9 El personal no debe usar joyas ni cosméticos en las áreas de producción.
7. Documentación legal y técnica
7.1 Aspectos generales.
7.1.1 Todos los documentos deben ser escritos en español, impresos en un medio que asegure su legibilidad, empleando vocabulario sencillo, indicando el tipo, naturaleza, propósito o uso del documento.
La organización de su contenido será tal que permita su fácil comprensión.
7.1.2 Los documentos deben ser emitidos a través de un método de reproducción que evite cualquier posibilidad de error durante la transcripción.
7.1.3 La documentación se debe archivar en forma tal que sea de fácil y rápido acceso.
7.1.4 Todos los documentos maestros deben incluir: título, tipo de documento, paginación, fecha de emisión, nombre, firma y posición dentro de la empresa de las personas que elaboraron, revisaron y autorizaron el documento.
7.1.5 Los originales de los documentos maestros que presenten modificaciones, se retendrán durante
5 años, después de su cancelación o sustitución.
7.1.6 Debe existir un sistema que permita la revisión, distribución y cualquier modificación o cancelación de un documento maestro. Dicho sistema debe incluir las instrucciones detalladas, el personal involucrado y definir las responsabilidades para asegurar la distribución de los documentos actualizados y el retiro de los obsoletos.
7.1.7 Los documentos destinados al registro de datos durante el proceso deben ser diseñados con suficiente espacio para los datos que habrán de registrarse.
7.1.8 La conservación de los registros de producción, acondicionamiento, control y distribución de
los lotes elaborados, debe ser de un año después de la fecha de caducidad del producto.
7.2 El establecimiento debe contar como mínimo con los siguientes documentos legales:
7.2.1 Licencia sanitaria o aviso de funcionamiento expedido por la SSA, según el caso.
7.2.2 Constancia de aviso del responsable sanitario.
7.2.3 Registro en el padrón ante la SECOFI.
7.2.4 Organigrama del establecimiento, indicando los puestos clave y las personas que los ocupan.
7.2.5 Edición vigente de la FEUM, así como los suplementos correspondientes.
7.2.6 Relación de medicamentos registrados.
7.2.7 Expediente legal de cada producto, el cual debe estar conformado por los siguientes documentos como mínimo:
7.2.7.1 Original del oficio de otorgamiento de registro emitido por la SSA.
7.2.7.2 Original de oficios de aprobación o rechazo a modificaciones que haya tenido el producto, emitidos por la SSA.
7.2.7.3 Proyectos de marbete para envases primarios, secundarios y etiquetas, autorizados por el departamento técnico de la SSA, para todas las presentaciones expresadas en el oficio de registro.
7.2.7.4 Cualquier otro oficio emitido por la SSA, con relación al producto.
7.2.8 Libro de control de estupefacientes y psicotrópicos, en su caso.
7.3 El establecimiento debe contar, como mínimo, con los siguientes documentos técnicos:
7.3.1 Planos actualizados del establecimiento, entre los cuales se deben incluir los de los sistemas críticos.
7.3.2 Relación del equipo de producción.
7.3.3 Relación de equipos e instrumentos analíticos.
7.3.4 El expediente maestro de cada producto, el cual debe estar conformado por los siguientes documentos:
7.3.4.1 Información sometida para la obtención del registro.
7.3.4.2 Información sometida para la solicitud de modificaciones a las condiciones originales del registro del producto.
7.3.4.3 Fórmula cualitativa-cuantitativa.
7.3.5 Orden maestra de producción para cada tamaño de lote, la cual debe incluir:
7.3.5.1 Nombre del producto, forma farmacéutica y concentración.
7.3.5.2 Relación completa de los componentes que intervienen en la elaboración del medicamento, aparezcan o no en el producto terminado, incluyendo clave, nombre y cantidad.
7.3.5.3 Instrucciones completas para la elaboración del producto, detallando equipo principal, parámetros críticos y precauciones a seguir.
7.3.5.4 Indicación de los rendimientos máximos y mínimos, tanto en etapas intermedias, como al final del proceso.
7.3.6 Orden maestra de acondicionamiento para cada presentación, y de acuerdo con el equipo de acondicionamiento, la cual debe contener la siguiente información:
7.3.6.1 Nombre del producto, forma farmacéutica y concentración.
7.3.6.2 Relación completa de los materiales para el acondicionamiento, incluyendo clave, nombre y cantidad.
7.3.6.3 Instrucciones detalladas para el proceso de acondicionamiento.
7.3.7 Especificaciones del producto.
7.3.8 Método analítico para el producto.
7.3.9 Especificaciones de materias primas, o referencia de las mismas utilizadas en el establecimiento.
7.3.10 Descripción de la presentación o presentaciones del producto y el tipo de envase primario y secundario.
7.3.11 Especificaciones para material de acondicionamiento.
7.3.12 PNO para limpieza y operación de los equipos utilizados en la fabricación de los productos.
7.3.13 PNO’s para la limpieza de las áreas de fabricación.
7.3.14 PNO’s para las operaciones relacionadas con los sistemas críticos del establecimiento.
7.4. Se debe contar con el expediente de cada lote elaborado, el cual debe contener como mínimo con:
7.4.1 Orden de producción de cada lote elaborado, mediante la cual pueda comprobarse que el producto fue fabricado e inspeccionado de acuerdo con los procedimientos y las instrucciones descritas en el expediente maestro.
7.4.2 Orden de acondicionamiento, mediante la cual pueda comprobarse que el producto fue revisado, identificado y acondicionado de acuerdo con lo establecido en los procedimientos e instrucciones del expediente maestro.
7.4.3 Reportes analíticos del producto en sus distintas etapas.
7.5 Se debe contar con los registros de los resultados analíticos de materias primas, material de acondicionamiento y producto terminado, así como la documentación que avale los resultados del laboratorio de control conforme a los requerimientos de las buenas prácticas de laboratorio.
7.6 Se debe contar con los registros de distribución que contengan, como mínimo, la siguiente información para cada lote de producto distribuido:
7.6.1 Nombre del producto.
7.6.2 Presentación.
7.6.3 Número de lote.
7.6.4 Identificación del cliente o receptor.
7.6.5 Cantidad enviada.
7.6.6 Fecha de envío y recibo.
7.7 Deben existir registros de quejas, que contengan toda la información relacionada con:
7.7.1 El motivo.
7.7.2 La revisión de las muestras y datos de la misma.
7.7.3 Los resultados de las investigaciones realizadas para cada una.
7.7.4 La determinación de las acciones correctivas y medidas adoptadas, así como el seguimiento respectivo, sobre la implementación de las mismas.
7.7.5 La determinación de la responsabilidad.
7.8 Deben existir registros de devoluciones, que contengan la siguiente información como mínimo para cada una:
7.8.1 Nombre del producto y presentación.
7.8.2 Cantidad devuelta.
7.8.3 Nombre y localización de quien devuelve.
7.8.4 Causa y dictamen técnico de la devolución.
7.8.5 Destino del producto y autorizaciones correspondientes.
8. Diseño y construcción de un establecimiento de la industria químico farmacéutica.
8.1 El establecimiento debe estar localizado, diseñado, construido y conservado de acuerdo con las operaciones que en él se efectúen. Su construcción y distribución deben asegurar la protección de los productos contra contaminación.
8.1.1 Debe colocarse en la entrada de la empresa, en la fachada, un rótulo donde se indique el nombre y clasificación del establecimiento, y otro que indique el nombre y número de autorización del responsable, el número de la cédula profesional, su horario de asistencia y el nombre de la institución superior que expidió el título profesional.
8.1.2 Debe permitir la seguridad y acceso controlado del personal a las áreas de producción, almacenes y control de calidad.
8.1.3 Debe existir un área de recepción y distribución que garantice la conservación de la calidad de los insumos y productos.
8.1.4 Las actividades de conservación deben ser programadas, realizadas y documentadas de tal manera que eviten los riesgos de contaminación al producto.
8.1.5 Las áreas de producción, almacenamiento y control de calidad no deben ser usadas como vías de acceso para el personal.
8.1.6 Debe contar con sistemas de descarga de aguas residuales. El sistema de descarga de aguas negras debe ser independiente del drenaje pluvial.
8.2 Las dimensiones de las diferentes áreas deben ser en función de la capacidad de producción, de la diversidad de productos y tipo de operaciones al que se destine cada una.
8.2.1 Se debe contar con áreas que posean el tamaño, diseño y construcción para efectuar los procesos de fabricación correspondientes, así como permitir un flujo de materiales y personal que no ponga en riesgo la calidad de los productos y procesos, y garantice su seguridad y eficiencia.
8.3 Las superficies interiores de las áreas de producción deben contar con acabados sanitarios.
8.4 Las instalaciones de ductos de ventilación, líneas de energía eléctrica y otros servicios inherentes a las áreas de producción deben encontrarse ocultas o fuera de éstas. Su ubicación y diseño debe ser tal, que permita su mantenimiento.
8.4.1 Las áreas deben estar iluminadas y ventiladas y contar, en caso de que así lo requieran, con control de aire, polvo, temperatura y humedad.
8.4.2 Los sistemas de ventilación y extracción de aire deben estar diseñados de tal forma que no permitan el ingreso de contaminantes externos.
8.4.3 Las lámparas de las áreas de producción deben estar diseñadas y construidas de tal forma que eviten la acumulación de polvo y permitan su limpieza. Deben contar con cubierta protectora lisa.
8.5 Las áreas de producción, acondicionamiento, y sus servicios inherentes (particularmente los sistemas de aire) de penicilínicos, cefalosporínicos, citotóxicos, inmunodepresores, hormonales, hemoderivados, biológicos virales, biológicos microbianos y otros considerados como de alto riesgo por la autoridad sanitaria, deben ser completamente independientes.
8.6 Las instalaciones destinadas para el manejo de animales de laboratorio deben encontrarse aisladas de las áreas de fabricación.
8.7 Los almacenes deben tener capacidad y condiciones de temperatura y humedad relativa requeridos para la conservación de materia prima, materiales y productos.
8.8 Las condiciones de trabajo (temperatura, vibraciones, humedad, ruido, polvo), no deben perjudicar al operador ni al producto, directa o indirectamente.
8.9 Las presiones diferenciales de aire de las áreas de producción deben estar balanceadas de tal forma que eviten cualquier tipo de contaminación.
8.10 Las áreas de producción deben contar con indicadores de presión diferencial.
8.11 Los pasillos internos de los módulos de producción deben contar con aire filtrado.
8.12 Las áreas de producción donde se generen polvos deben contar con sistemas de recolección y procedimientos para la disposición final de polvos colectados.
8.13 El diseño de los sistemas de extracción debe ser tal que evite una potencial contaminación cruzada.
8.14 Las tuberías fijas deben estar identificadas, en base al código de colores de la NOM-028-STPS-1994 "para servicios generales".
8.15 Si los drenajes están conectados directamente a una coladera o alcantarilla, deben tener una trampa o algún dispositivo que evite contaminación.
8.16 Cuando se requiera tener un canal, éste debe ser de fácil limpieza y sanitización.
8.17 Debe existir un área específica para efectuar las operaciones de acondicionamiento, que facilite el flujo de personal, materiales y productos.
8.18 El laboratorio de control de calidad debe estar separado físicamente de las áreas de producción y almacenes, contar con espacio e instalaciones para las pruebas y análisis que se realicen, existir separación física entre las áreas de análisis, instrumentos de medición, área de reactivos y pruebas microbiológicas.
8.19 Se debe contar con un área específica para las muestras de retención de los productos fabricados.
8.20 Las áreas destinadas para cambio y almacenamiento de ropa de trabajo, lavado, duchas y servicios sanitarios deben estar en lugares de fácil acceso y en correspondencia con el número de trabajadores.
Los servicios sanitarios no deben comunicarse directamente ni localizarse en vías de paso con las áreas de producción o almacenamiento y deben estar provistos de:
8.20.1 Ventilación.
8.20.2 Agua fría y caliente.
8.20.3 Lavabos.
8.20.4 Mingitorios e inodoros.
8.21 En caso de contar con comedor, éste debe estar separado de las áreas de fabricación.
8.22 Se debe contar con un área específica para el taller de mantenimiento separada de las otras áreas de fabricación.
8.23 Se debe contar con un área destinada al servicio médico, separada físicamente de las áreas de fabricación.
9. Control de la fabricación.
9.1. Generalidades.
9.1.1 El manejo de materia prima, materiales de acondicionamiento y productos debe seguir procedimientos e instrucciones escritas.
9.1.1.1 Debe contarse con un PNO para el manejo de las sustancias y productos que contengan estupefacientes y psicotrópicos, que considere los aspectos de la regulación sanitaria vigente.
9.1.2 Productos intermedios y productos a granel adquiridos como tales, deben ser manejados como si fueran materias primas.
9.1.3 En el manejo de materias primas y productos secos deben tomarse precauciones para controlar la generación y dispersión de polvos.
9.1.4 Al inicio y durante el proceso las materias primas, materiales de acondicionamiento, envases con producto a granel, equipos y áreas utilizadas, deben identificarse indicando el producto que se está elaborando, el número de lote y, cuando proceda, la fase de producción.
9.1.5 Las etiquetas de identificación de los envases, equipos o áreas, deben ser claras, inequívocas y de un formato aprobado.
9.1.6 El acceso a las áreas de fabricación queda limitado al personal autorizado.
9.1.7 Los PNO´s deben estar accesibles al personal involucrado.
9.1.8 El muestreo para el control del proceso debe llevarse a cabo en base a PNO´s.
9.1.9 El producto terminado en su empaque final, se considera en retención temporal hasta que sean efectuados todos sus análisis y sea liberado por control de calidad para su distribución.
9.1.10 Se debe contar con registros de humedad y temperatura, los cuales demuestren que las condiciones son adecuadas para el almacenamiento de las materias primas, material de acondicionamiento y productos.
9.1.11 En caso de que se requiera un mantenimiento correctivo del equipo durante la producción deben establecerse PNO´s para evitar la contaminación del producto.
9.2. Control de adquisición y recepción de materias primas y material de acondicionamiento.
9.2.1 Adquisición.
9.2.1.1 Las materias primas y material de acondicionamiento deben comprarse a proveedores aprobados, de conformidad con el sistema de control de calidad interno.
9.2.1.2 Debe realizarse en base a las especificaciones internas.
9.2.2 Recepción.
9.2.2.1 Al recibir cualquier envío, se debe verificar que los recipientes se encuentren identificados (nombre, cantidad y número de lote o equivalente), cerrados, que no presenten deterioro o daños de cualquier tipo que puedan afectar las características de calidad del material que contienen y que concuerde con lo indicado en la orden de compra y factura.
9.2.2.2 Al recibir cada lote de materia prima o material de acondicionamiento se debe asignar un número de lote interno.
9.2.2.3 Los recipientes se deben colocar sobre tarimas o anaqueles de tal manera que se facilite su limpieza, inspección y manejo.
9.3. Control del almacenamiento de materias primas, material de acondicionamiento y producto.
9.3.1 Debe realizarse con base en lo establecido en PNO´s que consideren la clara identificación y separación por medios físicos o sistemas de control.
9.3.2 Debe realizarse utilizando equipo que esté de acuerdo con sus características.
9.3.3 Se debe contar con PNO´s para la limpieza y mantenimiento de los almacenes.
9.3.4 Se debe contar con un PNO basado en el sistema de primeras entradas primeras salidas (PEPs).
9.3.5 Las materias primas, materiales de acondicionamiento y productos en cualquiera de sus etapas de fabricación, deben colocarse sobre tarimas.
9.3.6 Las materias primas y los materiales de acondicionamiento deben muestrearse de acuerdo con el PNO correspondiente, analizarse y dictaminarse antes de su uso. Los envases muestreados, deben indicarlo en su identificación.
9.3.7 Las materias primas y los materiales de acondicionamiento, cuya vigencia de aprobación ha terminado, deben ponerse en retención temporal, para su reanálisis y/o disposición final.
9.3.8 Las materias primas, material de acondicionamiento o productos rechazados deben ser identificados como tales y trasladados a una área específica delimitada, para evitar su uso en cualquier proceso productivo. Deben ser confinados, destruidos, devueltos o reprocesados, según dictamen, lo que debe quedar registrado.
9.3.9 Se debe contar con un programa para el control y erradicación de fauna nociva.
9.4. Preparación y surtido de materias primas y material de acondicionamiento.
9.4.1 Deben existir PNO´s que especifiquen como mínimo:
9.4.1.1 Que el manejo se realice sólo por personal autorizado.
9.4.1.2 Que asegure que son medidos, pesados y/o contados con exactitud. Estas operaciones deben ser verificadas y registradas por una segunda persona.
9.4.1.3 Medidas para evitar la contaminación cruzada.
9.4.1.4 El tipo de indumentaria que debe llevar el personal en función de las características de la materia prima y del área.
9.4.1.5 Que el surtido sea verificado y ambas operaciones registradas.
9.4.1.6 Que cada envase de materias primas o paquetes de material de acondicionamiento esté identificado con: nombre, cantidad, lote (interno), y nombre y lote del producto en que será utilizado.
9.4.1.7 Que se cancele la identificación y se controle la disposición posterior de los envases vacíos.
9.4.2 Las materias primas y materiales de acondicionamiento preparados para la producción y/o acondicionamiento deben mantenerse en una área destinada para ello, separados por lote de producto.
9.4.3 Los registros de inventario deben llevarse de tal manera que permitan la conciliación y rastreabilidad por lote de las cantidades recibidas contra las cantidades surtidas. En caso de existir discrepancias fuera de los límites establecidos, se debe emitir un reporte.
9.5. Control de producción.
9.5.1. Consideraciones generales.
9.5.1.1 Cada lote de producto se debe controlar mediante la orden de producción verificada por personal autorizado.
9.5.1.2 Cuando se requieran efectuar ajustes de la cantidad a surtir, en función de la potencia de las materias primas, deben calcularse y verificarse por personal autorizado y quedar documentados en la orden de producción.
9.5.1.3 La recepción de los materiales la debe realizar personal autorizado, quien después de verificar firmará en la orden.
9.5.1.4 La orden de producción correspondiente debe estar a la vista del personal que realiza el proceso antes y durante la producción.
9.5.1.5 El área de trabajo debe estar libre de materiales, documentos e identificaciones de lotes procesados con anterioridad o ajenos al lote que se va a procesar.
9.5.1.6 Antes de iniciar la producción, se debe autorizar el uso del área previa verificación y documentación de que el equipo y las áreas están limpios e identificados, de acuerdo con el PNO correspondiente.
9.5.1.7 El encargado del proceso debe verificar que el personal que intervenga en la producción use la indumentaria y los equipos de seguridad, de acuerdo con la orden de producción y/o al PNO correspondiente.
9.5.1.8 Las operaciones deben realizarse de acuerdo con la orden de producción y registrarse en la misma en el momento de llevarse a cabo.
9.5.1.9 La orden de producción debe indicar las operaciones que deben ser supervisadas.
9.5.1.10 La orden de producción debe precisar los parámetros y controles del proceso que sean requeridos.
9.5.1.11 Los resultados de las pruebas y/o análisis realizados durante el proceso, deben registrarse en la orden de producción o anexarse.
9.5.1.12 Los encargados de producción y del área de calidad deben revisar, documentar y evaluar cualquier desviación a la orden de producción y definir las acciones conducentes.
9.5.1.13 El rendimiento final y los rendimientos intermedios indicados en la orden de producción, deben ser registrados y comparados contra sus límites, en caso de excederlos se debe llevar a cabo una investigación y anexar el resultado de la misma en la orden de producción.
9.5.1.14 Deben existir PNO´s que garanticen la separación e identificación de los productos.
9.5.1.15 Deben realizarse controles durante el proceso que garanticen que el producto permanece dentro de especificaciones.
9.5.1.16 Debe revisarse la orden de producción, de acondicionamiento, los registros, resultados analíticos, etiquetas y demás documentación inherente. Comprobando que cumple con las especificaciones de proceso establecidas; sólo el área de calidad puede dictaminar (liberar o rechazar).
9.5.2. Control de la producción de formas farmacéuticas sólidas.
9.5.2.1 Los equipos en que se generen polvos, deben estar provistos de sistemas de extracción eficientes y situados e instalados de forma que se evite contaminación cruzada, en cubículos físicamente separados, a menos que todos sean utilizados para fabricar el mismo lote de producto.
9.5.2.2 La disposición de los polvos colectados debe realizarse en base a PNO´s y conforme a lo que establezcan las disposiciones aplicables. La limpieza de los colectores debe llevarse a cabo de acuerdo con el PNO correspondiente.
9.5.2.3 Debe contarse con un control que evite contaminación cruzada en las mangas y filtros de los secadores de lecho fluidizado. Para productos en que este control no sea suficiente, se debe emplear un juego de mangas y/o filtros exclusivos por producto.
9.5.2.4 Se debe contar con un registro del uso e inspección de tamices, dosificadores, punzones y matrices para detectar el momento en que no cumplen con las especificaciones.
9.5.2.5 Las cápsulas de gelatina dura vacías, deben ser consideradas y tratadas como una materia prima.
9.5.2.6 Control de la producción de formas farmacéuticas líquidas y semisólidas no estériles.
9.5.2.6.1 El área de producción debe contar con suficientes tanques y marmitas de preparación con tapa y cuando se requiera, enchaquetados y con sistemas de agitación.
9.5.2.6.2 Los tanques, recipientes, líneas de conducción y bombas deben ser diseñados, construidos e instalados de forma que puedan limpiarse y sanitizarse fácilmente.
9.5.2.6.3 Se debe contar con tomas identificadas de agua purificada.
9.5.2.6.4 Después de sanitizar los sistemas de agua por medios químicos debe seguirse un procedimiento validado a fin de garantizar que el agente sanitizante ha sido eliminado.
9.5.2.6.5 Las líneas de conducción por las que se transfieran materias primas o productos, deben ser de un material inerte que no contamine y estar identificadas.
9.5.2.6.6 Se debe garantizar la homogeneidad del producto durante las distintas fases del proceso.
9.5.2.6.7 Cuando el producto no se envase inmediatamente, se deben especificar sus condiciones y periodo máximo de almacenamiento.
9.5.2.6.8 Se deben mantener registros de las temperaturas de proceso en las etapas críticas del mismo.
9.5.2.6.9 Se deben llevar gráficas de control de peso o volumen durante el proceso de llenado y anexarlas a la orden de producción.
9.5.3. Control de la producción de formas farmacéuticas estériles.
9.5.3.1 La producción de formas farmacéuticas estériles debe realizarse en áreas limpias a las que el personal, el producto y/o los materiales ingresen o salgan cumpliendo con los requisitos que establezca el PNO correspondiente a fin de evitar contaminación.
9.5.3.2 Las áreas limpias deben mantenerse con el grado de limpieza que corresponda a su clasificación (ver anexo 1), recibiendo aire que haya pasado a través de filtros con el grado de eficiencia establecido en el diseño y construcción.
9.5.3.3 Las diversas operaciones de preparación de materiales y productos, llenado y esterilización, deben realizarse en zonas separadas dentro del área limpia.
9.5.3.4 Para productos que se procesen por técnica de llenado aséptico debe cumplirse con los parámetros que se establezcan en un protocolo de prueba de simulación de proceso.
9.5.3.5 Los procesos de esterilización deben estar validados.
9.5.3.6 En las áreas limpias debe estar presente el mínimo de personas necesarias; esto es especialmente importante durante los procesos asépticos, en cuyo caso y en la medida de lo posible, deben inspeccionarse y controlarse desde el exterior.
9.5.3.7 El personal empleado en estas áreas (incluyendo el de limpieza y el de mantenimiento) debe recibir capacitación en: conceptos básicos de microbiología, técnicas de vestido, técnicas asépticas, reglas de higiene y otros temas específicos para productos estériles.
9.5.3.8 El material y diseño de la ropa debe ser confortable y generar el mínimo de partículas. La utilizada en el área aséptica debe ser previamente esterilizada.
9.5.3.9 El sistema de aire debe controlarse de tal manera que cumpla con los parámetros de su diseño (flujo, velocidad, diferenciales de presión, cantidad de partículas, humedad, temperatura, biocarga y ruido).
9.5.3.10 Se debe contar con indicadores y/o alarmas para detectar oportunamente fallas en el sistema de aire, para tomar las medidas necesarias.
9.5.3.11 El equipo, los sistemas de aire, agua y esterilización, deben ser objeto de mantenimiento y calificación de manera periódica y documentada.
9.5.3.12 Deben tomarse en cuenta los resultados del control ambiental durante las operaciones asépticas, para dictaminar un lote, como complemento al resultado analítico final.
9.5.3.13 Deben existir PNO’s que establezcan tiempo límite entre:
- la esterilización y la utilización de los materiales,
- la preparación y esterilización/llenado del producto,
- la recolección de agua grado inyectable y su uso,
- el inicio y término del llenado.
- tiempo de permanencia del personal dentro de las áreas involucradas.
9.5.3.14 Después del llenado, los productos parenterales deben inspeccionarse para la detección de partículas y otros defectos de acuerdo con un PNO.
9.5.3.15 Los operarios que realicen la inspección para el control de partículas de productos estériles deben someterse a controles periódicos de agudeza visual.
9.5.3.16 Se debe realizar la prueba de hermeticidad a los productos parenterales de acuerdo con un PNO.
9.6. Control del acondicionamiento.
9.6.1. Consideraciones generales.
9.6.1.1 Cada lote de producto se debe controlar mediante una orden de acondicionamiento, verificada por personal autorizado.
9.6.1.2 Deben existir áreas específicas para el acondicionamiento para evitar confusiones y mezclas de los materiales y/o productos.
9.6.1.3 Antes de iniciar el acondicionamiento, se debe verificar que el equipo y las áreas estén limpios, libres de materiales ajenos e identificados conforme a PNO´s.
9.6.1.4 El personal que intervenga en el acondicionamiento, debe usar la indumentaria indicada en el PNO respectivo.
9.6.1.5 Las operaciones de acondicionamiento se deben realizar de acuerdo con la orden respectiva y registrarse conforme se vayan llevando a cabo.
9.6.1.6 En la orden de acondicionamiento se deben indicar las operaciones que deben supervisarse.
9.6.1.7 Los controles que se lleven a cabo se deben indicar en la orden de acondicionamiento.
9.6.1.8 Los resultados de las pruebas realizadas durante el acondicionamiento se deben registrar y anexar en la orden respectiva.
9.6.1.9 Los encargados de acondicionamiento y del área de calidad deberán revisar, documentar y evaluar cualquier desviación a la orden de acondicionamiento y definir las acciones conducentes.
9.6.1.10 Al finalizar las operaciones de acondicionamiento se debe documentar el balance de los materiales y del producto. Las devoluciones, destrucciones y/o entrega de productos que procedan se deben realizar de acuerdo con PNO´s.
9.6.1.11 La devolución de material impreso debe documentarse. Esta información complementa el expediente del producto.
9.6.1.12 El rendimiento final y los rendimientos intermedios indicados en la orden de producción, deben ser registrados y comparados contra sus límites; en caso de excederlos se debe llevar a cabo una investigación y anexar el resultado de la misma en la orden de producción.
9.6.1.13 Debe revisarse la orden de producción, de acondicionamiento, los registros, resultados analíticos, etiquetas y demás documentación inherente. Comprobando que cumple con las especificaciones de proceso establecidas; sólo el área de calidad puede dictaminar (liberar o rechazar).
9.6.2. Control de la rotulación.
9.6.2.1 Deben existir áreas específicas para la rotulación de los materiales, que permitan evitar confusiones y mezclas.
9.6.2.2 La rotulación de los materiales debe ser revisada y verificada por personal autorizado, quien
debe registrarlo.
9.6.2.3 Deben existir PNO´s que garanticen la seguridad en el manejo de los materiales a rotular, de los rotulados, de los materiales para impresión y de los obsoletos.
9.6.2.4 Debe existir una área específica para el almacenamiento de materiales rotulados, si procede.
9.7. Maquilas
9.7.1 Las responsabilidades entre el maquilador y el contratante deben estar claramente establecidas en un contrato que debe contener, además de los aspectos comerciales, las especificaciones de proceso, producto y rendimiento.
9.7.2 El contratante debe notificar por escrito a la SSA las partes del proceso a realizar por el maquilador.
9.7.3 En el contrato de maquila el contratante asume la responsabilidad de la calidad del producto.
9.7.4 El maquilador debe proporcionar al contratante la documentación original referente a la fabricación del producto maquilado.
9.7.5 El contratante debe tener la posibilidad de supervisar la fabricación de su producto.
9.7.6 El contratante debe asegurar que los productos o materiales enviados por el maquilador se ajusten a las especificaciones.
9.8. Control de la distribución.
9.8.1 Deben establecerse PNO´s para el control de la distribución de los productos.
9.8.2 El sistema de distribución de los medicamentos debe establecerse de acuerdo con la política de primeras entradas primeras salidas (PEPs).
9.8.3 Debe garantizarse la identificación e integridad de los productos.
9.8.4 Los productos se deben manejar en condiciones de temperatura y humedad de acuerdo con lo establecido en la etiqueta.
9.8.5 Debe mantenerse un registro de distribución de cada lote de producto para facilitar su retiro del mercado en caso necesario.
9.8.6 Devoluciones y quejas.
9.8.6.1 Debe existir un PNO para el control de los productos devueltos que considere como mínimo:
9.8.6.1.1 Que deben ponerse en retención temporal y ser evaluados por control de calidad para determinar si deben liberarse, reprocesarse o destruirse.
9.8.6.1.2 Registros de recepción, evaluación y destino.
9.8.6.2 Debe existir un PNO para el manejo de quejas que considere como mínimo:
9.8.6.2.1 La obligatoriedad de la atención de todas las quejas.
9.8.6.2.2 La necesidad de identificar la causa de la queja.
9.8.6.2.3 Definición de las actividades a realizar respecto al problema.
9.8.6.2.4 Los casos que se requieran notificar a la autoridad sanitaria y la forma de hacerlo.
9.8.6.2.5 La forma de notificar al cliente, en su caso.
9.8.6.2.6 Los registros de los resultados obtenidos y las decisiones tomadas en relación a las quejas.
9.9. Control de recuperación o reproceso de materiales.
9.9.1 Debe elaborarse un PNO para la recuperación y/o reproceso de productos.
9.9.2 La recuperación y/o el reproceso deben llevarse a cabo con la revisión y autorización previa del área de calidad.
9.9.3 Debe elaborarse una orden de producción y/o acondicionamiento específica para el lote a recuperar y/o reprocesar, la cual debe ser autorizada por el responsable sanitario.
9.9.4 La liberación de un lote reprocesado debe contar con la autorización del responsable sanitario.
9.10. Control de la contaminación.
9.10.1 Las áreas utilizadas para la producción y acondicionamiento deben estar separadas y comunicarse entre sí de acuerdo con un orden que corresponda a la secuencia de las operaciones y a los niveles de limpieza requeridos, de forma que se minimice el riesgo de confusión, se evite la contaminación cruzada y se disminuya el riesgo de omisión o ejecución errónea de cualquier fase de la fabricación.
9.10.2 Se debe contar con sistemas de inyección y extracción de aire filtrado en las áreas de producción y acondicionamiento, que eviten contaminación cruzada; la contaminación por el ambiente externo y la contaminación ambiental.
9.10.3 El acceso a las áreas de producción y acondicionamiento debe ser restringido y definido por PNO´s que indiquen la indumentaria requerida y su uso para cada área.
9.10.4 Las áreas y equipos deben limpiarse y sanitizarse de acuerdo con PNO´s específicos que aseguren la disminución de microorganismos y otros contaminantes a límites preestablecidos.
9.10.5 Se deben realizar evaluaciones periódicas para verificar que los límites de contaminación microbiológica en áreas y superficies, se mantienen dentro de lo establecido.
9.11. Validación
9.11.1 Los procesos de producción deben ser validados en base a protocolos que tomen en cuenta los aspectos de:
9.11.2 Personal, áreas, materias primas, equipo y sistemas generales. El grado y alcance del trabajo de validación dependerá de la naturaleza y complejidad del producto y proceso involucrado.
9.11.3 Los métodos analíticos deben ser validados, de acuerdo con lo establecido en el apartado 9.12 "control del laboratorio analítico".
9.11.4 Los sistemas críticos y equipos de producción y acondicionamiento deben ser calificados de acuerdo con protocolos que tomen en cuenta su diseño, construcción, instalación y operación.
9.11.5 La documentación relativa a los estudios de validación debe estar completa, ordenada y disponible.
9.11.6 Debe existir un sistema de control de cambios que regule las modificaciones que puedan afectar la calidad del producto y/o la reproducibilidad del proceso, método o sistema.
9.11.7 Los procesos deben ser objeto de revalidación en base a políticas que establezca la empresa, para garantizar que siguen siendo capaces de proporcionar los resultados previstos.
9.12. Control del laboratorio analítico.
9.12.1 Se debe contar con especificaciones escritas para la evaluación de cada lote de materia prima, producto a granel, material de acondicionamiento y producto terminado.
9.12.2 Se debe contar con PNO´s para el muestreo de materia prima, producto a granel, material de acondicionamiento y producto terminado.
9.12.3 Se debe contar con métodos de análisis validados para producto a granel, producto terminado y materia prima en caso de no aparecer en cualquier farmacopea internacional ni en la FEUM.
9.12.4 Se debe contar con métodos de prueba para el material de acondicionamiento.
9.12.5 Se debe contar con un programa de calibración de instrumentos de medición.
9.12.6 Se deben realizar los estudios de estabilidad, de acuerdo con lo requerido por la
NOM-073-SSA1-1993.
9.12.7 Deben conservarse muestras de retención representativas de cada lote de producto, así como de cada uno de los materiales involucrados en la fabricación de éste.
El tiempo de retención debe ser de cuando menos 1 año después de su fecha de caducidad.
9.12.8 Deben existir PNO’s para la limpieza, mantenimiento y operación de cada uno de los instrumentos y equipos del laboratorio analítico que contemplen los registros correspondientes.
9.12.9 Los reactivos deben prepararse de acuerdo con la FEUM y suplementos vigentes. En caso de que en ésta no aparezca la información, podrá recurrirse a farmacopeas de otros países cuyos procedimientos de análisis se realicen conforme a especificaciones de organismos especializados u otra bibliografía científica reconocida internacionalmente.
9.12.10 La etiqueta de los reactivos debe indicar como mínimo: nombre, fecha de preparación, nombre de quien lo preparó, referencia de su registro, concentración, factor de valoración, caducidad, condiciones de almacenamiento, fecha de revaloración y fecha de recepción cuando se compren preparados.
9.12.11 Las sustancias de referencia primarias y secundarias deben fecharse, almacenarse, manejarse y utilizarse de manera que no se afecte su calidad. Se debe registrar el origen, identidad, cualquier información relativa a su preparación y caracterización, la fecha en que se usa y su vida útil.
9.12.12 Los medios de cultivo deben prepararse de acuerdo con la FEUM y suplementos vigentes.
En caso de que en ésta no aparezca la información, podrá recurrirse a farmacopeas de otros países cuyos procedimientos de análisis se realicen conforme a especificaciones de organismos especializados u otra bibliografía científica reconocida internacionalmente.
9.12.12.1 Deben utilizarse controles negativos y positivos como testigos durante el uso de los medios de cultivo.
10. Equipo de fabricación
10.1 Se debe contar con el equipo que posea el diseño y tamaño correspondientes a los procesos de fabricación, y estar localizado de manera tal que facilite su operación, limpieza y mantenimiento.
10.2 El equipo debe estar construido de tal forma que facilite su desmontaje, limpieza, montaje y mantenimiento.
10.2.1 El equipo debe estar construido de tal manera que las superficies en contacto con los componentes de la fórmula, los materiales del proceso o los productos, no sean reactivas, aditivas o absortivas como para alterar la seguridad, identidad, potencia, calidad o pureza del producto.
10.2.2 Cualquier sustancia requerida para la operación del equipo, como lubricantes, refrigerantes u otros, no deben estar en contacto con los componentes de la fórmula, envases primarios del producto o del producto en sí.
10.2.3 Los accesorios como tanques y tolvas deben contar con cubiertas.
10.2.4 Los engranajes y partes móviles deben estar protegidos para evitar la contaminación del producto en proceso y por seguridad del operario.
10.3 Limpieza y mantenimiento del equipo.
10.3.1 El equipo y los utensilios deben limpiarse, mantenerse y sanitizarse de acuerdo con un programa establecido para prevenir el mal funcionamiento o contaminación.
10.3.2 Se debe retirar toda la documentación y etiquetas, adheridas al equipo que identifique al lote del proceso anterior.
10.3.3 Deben existir PNO's de limpieza y mantenimiento escritos y aplicarse para la limpieza y mantenimiento del equipo y utensilios usados durante la producción, envasado o manejo del producto.
10.3.4 El equipo debe permanecer limpio, protegido e identificado cuando no se esté utilizando.
10.3.5 Se debe verificar la limpieza del equipo antes de ser utilizado.
10.3.6 El equipo debe estar calificado para el producto que se va a fabricar.
10.4 Todo equipo utilizado en la producción, empaque o manejo de los productos debe encontrarse localizado e instalado de tal manera que:
10.4.1 No obstaculice los movimientos del personal y facilite el flujo de los materiales.
10.4.2 Se asegure el orden durante los procesos y se controle el riesgo de confusión u omisión de alguna etapa del proceso.
10.4.3 Permita su limpieza y la de las áreas adyacentes, y no interfiera con otras operaciones del proceso.
10.4.4 Esté físicamente separado y, cuando sea necesario, aislado de cualquier otro equipo para evitar el congestionamiento de las áreas de producción, así como la posibilidad de contaminación cruzada.
10.5 Equipo automático, mecánico y electrónico.
10.5.1 Deben ser calibrados e inspeccionados, de acuerdo con un programa escrito diseñado para asegurar su funcionamiento. Las operaciones de calibración e inspección deben documentarse.
10.5.2 Con el fin de asegurar la exactitud de los datos manejados por estos sistemas, se debe implementar un sistema de protección de los mismos para evitar modificaciones a las fórmulas o registros efectuadas por personal no autorizado.
10.5.3 Se debe mantener un respaldo en copias fieles, cintas o microfilms, de toda la información archivada en las computadoras o los sistemas relacionados, para asegurar que la información emitida por estos sistemas es exacta, completa y que no existen modificaciones inadvertidas.
10.6 Filtros.
10.6.1 Los filtros empleados en la producción o el envasado de productos deben ser de materiales que no liberen fibras u otros cuerpos extraños. No se permite el uso de filtros de asbesto.
10.6.2 Si es necesario el uso de prefiltros que liberen fibras, posteriormente se debe filtrar la solución a través de un filtro que las retenga.
10.6.3 Los filtros deben ser compatibles con el producto a filtrar.
10.6.4 Debe efectuarse la prueba de integridad a los filtros utilizados en la esterilización de una solución, antes y después de este proceso. Estas operaciones deben verificarse y documentarse.
11. Destrucción y disposición final de residuos
11.1 Se debe contar con un sistema documentado que garantice el cumplimiento de las disposiciones legales en materia ecológica para la disposición final de residuos.
11.2 Se debe dar aviso a las autoridades competentes para llevar a cabo la disposición final de los mismos.
12. Concordancia con normas internacionales y mexicanas
Esta norma es parcialmente equivalente a los estándares internacionales:
ISO-8402: 1986 "Quality-vocabulary".
ISO 9000: 1987 "Quality management and quality assurance standards-guidelines for selection and use"
ISO 9001: 1987 "Quality systems-model for quality assurance in design/development, production, installation and servicing".
ISO 9002: 1987 "Quality systems-model for quality assurance in production and installation".
ISO 9003: 1987 "Quality systems-model for quality assurance in final inspection and test".
ISO 9004: 1987 "Quality management and quality systems elements-guidelines".
ISO 10011-1:1990 "Guidelines for auditing quality system- part 1: auditing".
ISO 10011-2: 1991 "Guidelines for auditing quality system-part 2: qualification criteria for quality systems auditors".
ISO 10011-3:1991 "Guidelines for auditing quality system-part 3: management of audit programmes".
Asimismo, es parcialmente equivalente a las normas mexicanas señaladas en Bibliografía.
13. Bibliografía
13.1 ANSI/ASQC 01-1988. Generic guidelines for auditing of quality systems.
13.2 CFR 1996 (Code of Federal Regulations), Title 21; Part 58, Good Laboratory Practice for Nonclinical Laboratory Studies. Washington, D.C., Office of Federal Register.
13.3 Evaluación y validación de sistemas críticos en áreas asépticas, Asociación Farmacéutica Politécnica, A.C. (1992).
13.4 Farmacopea de los Estados Unidos Mexicanos, 6a. Ed. México (1994).
13.5 Suplemento 1 Farmacopea de los Estados Unidos Mexicanos, sexta edición, México (1995).
13.6 Glosario de términos especializados para la evaluación de medicamentos. Versión preliminar (octubre 1990) (OMS).
13.7 Guías de prácticas adecuadas de fabricación para cuartos limpios. Monografía técnica No. 1 México (1992).
13.8 Guías generales de validación. SSA (1989).
13.9 Ley General de Salud (1997).
13.10 Ley General del Equilibrio Ecológico y la Protección al Ambiente (1996).
13.11 NMX-CC-1-1990 Sistemas de Calidad Vocabulario.
13.12 NMX-CC-2-1990 Sistemas de Calidad-Gestión de Calidad. Guía para la Selección, el Uso de Normas de Aseguramiento de Calidad.
13.13 NMX-Z-13 Guía para la Redacción, Estructuración y Presentación de las normas oficiales mexicanas.
13.14 NTE-CRP-001/88 Criterios para la determinación de residuos peligrosos y listado de los mismos.
13.15 Reglamento de Insumos para la Salud (1998).
13.16 Supporting and Suplementary Guidelines (OMS) 1992.
14. Observancia de la norma
Esta norma oficial mexicana es de observancia obligatoria en los establecimientos dedicados al proceso de los medicamentos y/o productos biológicos comercializados en el país, con el objeto de proporcionar medicamentos de calidad al consumidor.
Corresponde a la SSA, a través de la DGIS, la vigilancia del cumplimiento de las disposiciones de la presente norma, cuyo personal realizará los trabajos de verificación necesarios.
15. Anexos
I. CLASIFICACION DE AREAS
|
PARTICULAS |
CLASE 100
AREA CRITICA ASEPTICA 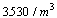
(BAJO FLUJO UNIDIRECCIONAL)  |
|
NO
VIABLES |
CLASE 10,000
AREA CRITICA ASEPTICA 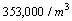
(FUERA DE FLUJO UNIDIRECCIONAL) |
|
partículas de 0.5 micras
y mayores |
CLASE 100,000
(AREA LIMPIA) 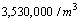 |
|
PARTICULAS |
CLASE 100
AREA CRITICA ASEPTICA 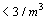
( BAJO FLUJO UNIDIRECCIONAL) |
|
VIABLES |
CLASE 10,000
AREA ASEPTICA
(FUERA DE FLUJO UNIDIRECCIONAL) 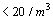 |
|
|
CLASE 100,000
(AREA LIMPIA) 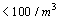 |
|
TEMPERATURA |
18 - 23º C |
|
HUMEDAD RELATIVA |
30 - 60 % * |
|
CAMBIOS DE
AIRE / HORA |
NO MENOS DE 20 |
|
VELOCIDAD DE FLUJO DE AIRE EN AREA CRITICA ASEPTICA (BAJO FLUJO UNIDIRECCIONAL) |
27 m/min ± 20 % ** |
|
PRESION |
NO MENOS DE 0.05 CM DE COLUMNA DE AGUA ENTRE AREAS ASEPTICAS |
|
DIFERENCIAL |
NO MENOS DE 0.12 CM DE COLUMNA DE AGUA ENTRE AREA ASEPTICA Y NO ASEPTICA |
* O MENOR CUANDO LAS CARACTERISTICAS DEL PRODUCTO EN PROCESO LO REQUIERA
** O MAYOR CUANDO LAS CARACTERISTICAS DEL PRODUCTO, PROCESO O AREA, LO REQUIERA.
16. Vigencia
La presente norma entrará en vigor con carácter obligatorio a los 180 días de su publicación en el Diario Oficial de la Federación.
Atentamente
Sufragio Efectivo. No Reelección.
México, D.F., a 15 de junio de 1998.- El Director General de Insumos para la Salud, Francisco J. Higuera Ramírez.- Rúbrica.
Fecha de publicación: 31 de julio de 1998
Si quiere obtener una copia del texto completo, presione aquí 